
This report was prepared by Clifford Goodman, Roy Ahn, Rick Harwood, Deborah Ringel, Kareen Savage, Daniel Mendelson, and Robert Rubin of The Lewin Group. Other staff that contributed to the report included Ansari Ameen, Dawn Bartoszewicz, Timothy Field, Doug Fountain, and George Steinfels of The Lewin Group. This report was prepared under contract for the Office of Health Policy, Office of the Assistant Secretary for Planning and Evaluation, Department of Health and Human Services (202-690-6870). The content of this report is solely the responsibility of The Lewin Group. For more information, please contact Clifford Goodman at 703-218-5500.
"Executive Summary
Purpose
The purpose of this project was to produce an analysis of the market barriers to the development of pharmacotherapies for substance abuse and addiction, and for cocaine abuse and addiction in particular. The analysis is intended to provide ASPE with information related to four areas:
- The characteristics of the market for substance abuse pharmacotherapies;
- Real and perceived market barriers;
- Case studies of pharmaceutical companies that have developed and marketed substance abuse pharmacotherapies (e.g., LAAM, naltrexone); and
- Industry's perception of the readiness of the science base.
This project emphasized the market for pharmacotherapies for cocaine abuse and addiction in order to focus the market analysis, interviews with industry representatives, and scenarios of new pharmacotherapy development subject to the resources available for the project. Nevertheless, the findings of these inquiries, along with the case studies, description of market barriers, and principal conclusions of this report, have direct relevance to markets for pharmacotherapies for substance abuse and addiction more broadly.
Principal Conclusions
Under current conditions, pursuing development of a new cocaine pharmacotherapy via a typical full product development cycle is not economically viable from the standpoint of industry. Although a variety of hurdles or procedural impediments may affect prospects for new pharmacotherapy development in this area, most of these are regarded as surmountable by the industry. However, three critical market barriers to significant progress in bringing an effective pharmacotherapy to a viable market are:
- Small and uncertain market for cocaine addiction and abuse pharmacotherapy;
- A substance abuse treatment system that limits access to this market; and
- Limited and uncertain payment for pharmacotherapy for this indication.
Methods
This study relied on several types of sources for information about cocaine pharmacotherapies and the market barriers to their development. These included:
- Analysis of the current market for cocaine abuse pharmacotherapies;
- Quantitative modeling of multiple hypothetical scenarios of pharmacotherapy development;
- Case studies of four selected pharmacotherapies; and
- Detailed interviews with executives of several private firms (e.g., pharmaceutical companies, venture capital firms) with real or potential interest in substance abuse and/or other central nervous system drug development.
The numbers of case studies and industry interviews were expressly limited by the scope of this project. Therefore, the information gathered from these sources cannot be considered to be systematically representative of pharmacotherapy experience or industry opinions pertaining to the market for substance abuse pharmacotherapies.
Principal Findings
Estimate of Cocaine Users in the U.S.
There are more than 2 million addicted or "heavy" cocaine users. Of these, as many as 800,000 to 900,000 may enter treatment at least once in a given year. On any given day, roughly 250,000 cocaine abusers are enrolled in treatment (i.e., who are at a residential facility or have been served at an ambulatory treatment center within the previous 30 days). Thus, of all heavy cocaine users, slightly more than 10 percent are enrolled in treatment on any given day.
Of the estimated 250,000 cocaine abusers, about 150,000 are primary cocaine abusers and about 100,000 are secondary cocaine abusers, i.e., who abuse cocaine secondarily to alcohol or opiates. A national survey data set (Treatment Episode Data Set 1992-1995), indicates that as many as 170,000 daily patients enrolled in treatment are secondary cocaine users. However, because cocaine abuse may be the third or fourth drug problem for many of these patients, a more conservative estimate of 100,000 secondary cocaine abusers is used. This brings the combined estimate of primary and secondary cocaine abusers to 250,000, which is more consistent with other recent estimates of between 200,000 and 250,000 daily cocaine patients in treatment. Indeed, estimates derived from the Drug Services Research Surveys for 1990 indicated there were 210,000 daily cocaine abusers in treatment, only a third of which were primary cocaine abusers.
It is estimated that there are approximately 11,500 centers providing treatment for cocaine abuse. Total spending on treatment exceeds $2 billion, and spending on treatment for cocaine abuse averages about $23.00 per patient enrollment day (including inpatients and outpatients), with spending of $9.00 per day for non-intensive outpatients.
Substance abuse treatment generally emphasizes a psychosocial, rather than medical, model. The most recent surveys that have examined staffing patterns confirm that the substance abuse treatment system involves little or no physician time in the treatment of patients. Even when on staff, physicians are often addressing primary health care needs or other mental disorders, rather than delivering specialized substance abuse treatment services.
Involvement of private practice physicians in treating cocaine addiction is virtually negligible. Of national expenditures for all specialized substance abuse services, including alcoholism, less than 1 percent are for psychiatrist visits. Of all national expenditures for substance abuse services excluding alcoholism, 2 to 3 percent are for visits to physicians of any type.
Expectations of Low Market Penetration
The proportion of the entire population of heavy substance abusers that is considered to be a realistic target market for pharmacotherapy is small relative to the target market of medications for other diseases. Although there are other clinical conditions for which the market penetration of medications is proportionately low, the absolute magnitude of an overall market may be so large that even low penetration of a substantially priced drug can be financially attractive to industry, as in the case of the smoking cessation market.
A combination of factors minimizes the attractiveness of the cocaine abuse market to industry, including: a relatively modest potential market (2.1 million heavy users), low proportion of users currently in treatment (250,000 enrollees on any given day), concerns about compliance in this population, and apparent market expectation of a low price point. This appears to be corroborated by the methadone market, where a relatively effective, low-priced medication in a well-established, long-standing treatment system achieves, at best, a 25 percent penetration of the small population of opiate addicts (i.e., about 125,000 patients enrolled in methadone treatment per day out of about 500,000 opiate addicts). The penetration of LAAM is less than five percent of the methadone market, i.e., less than one percent of the opiate addiction market, and the use of naltrexone for opiate addiction falls below that of LAAM.
Drawing inferences about the potential market for a new cocaine abuse medication from the market experience of other medications for substance abuse must consider the market-limiting characteristics of the treatment systems for opiate addiction to which methadone and LAAM are subject. The market conditions for a new cocaine abuse medication would differ if it is provided via more traditional means of physician prescribing and distribution through pharmacies, rather than if it is a Schedule II or Schedule III controlled substance warranting the forms of controlled treatment delivery required for methadone and LAAM. Nevertheless, their status as medications for illegal substance abuse, similarities in the user populations, and other characteristics make methadone, LAAM, and naltrexone useful, though imperfect, market comparators for a new cocaine abuse medication. As a group, the markets for these medications more closely resemble the potential market for a cocaine abuse mediation than markets for other medications, and industry looks to these markets accordingly when assessing the market for potential cocaine abuse medications.
The uncertainty about the number of cocaine users, especially the secondary users, places a wide confidence interval around the potential size of the market for cocaine abuse treatment. For the purposes of gauging the potential market for a new pharmacotherapy for cocaine addiction, it may be optimistic to use an estimate of 250,000 current daily enrollees in treatment. Projections of market penetration must consider that some primary users and many secondary abusers may be treated for other addictions and with behavioral therapy to the exclusion of pharmacotherapy, particularly in the context of the current treatment system. Given that many cocaine addicts abuse multiple substances and have diverse health and behavioral disorders, it may be that one or a few medications for cocaine abuse will be insufficient for treating this population. To the extent that multiple medications are needed, the market potential for any one medication would be reduced.
Basic Relationships of Price, Market Size, and Revenues
Pharmaceutical companies' decisions to pursue new products are based largely on financial considerations inherent in risk-reward tradeoffs. Financial indicators such as product net present value (NPV) and peak annual revenue (PAR) usually drive investment decisions. Beyond the appreciable risk associated with developing and marketing any new medication, the risk associated with developing and marketing a substance abuse pharmacotherapy is regarded by industry as quite considerable. (NPV is the difference between the present value of all cash inflows from a project and the present value of all cash outflows required for the investment, using an appropriate discount rate or required rate of return to calculate present values. PAR is the highest annual revenue achieved by a product during its market life.)
In principle, companies pursue projects that have positive NPVs. In addition, larger companies are less likely to be interested in pursuing a new drug for which PAR is projected to be less than $200 - $300 million. Of course, alternative projects that offer higher NPVs and PARs tend to be more attractive. This report examines relationships among price, market size, and revenues as they might affect PAR, NPV, and other indicators.
If a new medication were to be used by all 250,000 patients currently enrolled in treatment on any given day, it would have to sell at just over $2.00 per day in order to generate $200 million in PAR, a modest target PAR for most large pharmaceutical companies. If the greatest market penetration reached just 50 percent of currently enrolled patients, the price would have to be more than $4.00 per day. In order to achieve $200 million at a wholesale price of $0.50 per day, there would have to be more than 1 million daily patients.
Relative to current market conditions, optimistic assumptions are required to project a PAR that is comfortably in the target range for the larger pharmaceutical companies, which may be $300 500 million. For example, a market of 500,000 daily cocaine users in treatment (double the current level of 250,000) and retail prices of $2.75 - $5.00 (based on $2.50 wholesale) would achieve a PAR of $455 million. Such requirements would entail payments that would have to be realized in the form of new funding, reallocation of funds from the $2.1 billion currently spent for treatment of cocaine abuse, or a combination of these.
As no approved pharmacotherapy for cocaine abuse has been tested on the market, it is not possible to gauge directly the price sensitivity of that market. However, indirect available evidence from other substance abuse medications and the current nature of cocaine abuse treatment and its financing would indicate that the market would be very sensitive to the price of a cocaine medication. In a market where the average treatment cost is $9.00 per day for non-intensive outpatients, who constitute the great majority of all cocaine abuse patients, a cocaine pharmacotherapy priced at a daily dose of a few dollars would represent a significant proportionate cost increase. This may be particularly so in the estimation of substance abuse treatment providers that are vested in psychosocial approaches to the exclusion of pharmacotherapy. It is important to note that the price sensitivity of the current treatment system may vary considerably from that of more typical pharmacotherapy markets that involve physician prescribing and distribution through pharmacies.
The price of methadone may exert some pull on the price point for a cocaine medication. The price for that relatively effective medication, which is used to treat another stigmatized substance abuse population and is paid for primarily by government sources, is a mere 50 cents per daily dose. Although the price of medications for smoking cessation is considerably higher, payment for those medications does not come primarily from public sources, but rather by a self-pay population. The considerable price sensitivity of treatment programs subject to annual government appropriations has contributed to the disappointing market experience of two substance abuse medications, LAAM and naltrexone, which are priced higher than methadone, yet modestly priced compared to many prescription medications. While a price point that would be palatable to the current treatment system might be economically feasible for a company with a medication used by a very large market, it would not be feasible in the current market for cocaine abuse treatment.
Translating Market Barriers and Policy Options into Financial Parameters
Pharmaceutical companies' decisions to pursue new products are based largely on financial considerations inherent in risk-reward tradeoffs. Despite their diversity, most market barriers can be interpreted as having a direct effect on one or more financial parameters that are factored into these decisions. Similarly, most policy options that exist or that could be implemented to lower market barriers can be interpreted as having a direct effect on these financial parameters.
This report portrays relationships between market barriers and policy options, including those identified in the 1995 Institute of Medicine (IOM) report, Development of Medications for the Treatment of Opiate and Cocaine Addictions, and six basic parameters relevant to decisions to pursue a new therapy: R&D costs, time to product launch, marketing and distribution costs, market size/penetration, price, and duration of market life. These basic relationships were used in part to develop the scenarios for this study, and can be represented in quantitative modeling that generates such financial indicators as NPV and PAR.
Scenarios of Company Decision Making
This report presents several scenarios of pharmaceutical company decision making regarding whether to undertake projects to develop pharmacotherapies for cocaine addiction under various sets of market conditions. Using a quantitative model developed by The Lewin Group, the market conditions were translated into financial and other parameters to generate projected PAR and NPV for each scenario and for certain variations of these scenarios. Modeling these scenarios illustrates some of the key barriers and other limitations to development of medications for cocaine abuse, as well as how certain types of financial and policy options might reduce such barriers. Policy options to lower barriers that are used in these scenarios include some that already exist and some that have been posed by the IOM (1995).
The "Big Pharm Cold Start" scenario indicates that the prospects of developing a new medication for cocaine abuse and taking it through a full product development cycle do not appear favorable given a moderate wholesale price and an optimistic target market (i.e., 50 percent of the estimated 250,000 people currently enrolled in treatment for cocaine abuse). In order to achieve financial indicators that are more in line with traditional targets of large companies (e.g., PAR of $300 million) in this scenario, a considerable increase in price and/or market penetration would have to be realized. On the other hand, a modest and perhaps more realistic penetration of this market of 30,000 to 40,000 patients per day could also yield an acceptable PAR if a cocaine medication were priced at the premium levels (e.g., $25 - $30 per day) that are afforded triple pharmacotherapy for HIV/AIDS.
The "Biotech Gets Help" scenario suggests that, even for a company that is confident that it can develop a highly promising molecule with a relatively modest level of R&D expenditures and somewhat lower targets for financial performance, some combination of additional incentives may be needed. This scenario considers the impact of three government interventions: (a) regulatory reform that would shorten the time to launch by 1 year, (b) provision of market protection similar to orphan drug status, and (c) a significant commitment to expand treatment and financing capabilities at the state level. In this scenario (in which orphan-like status accords R&D tax breaks but no additional market protection because of existing patent protection), the regulatory reform and market protection have modest impacts compared to the expansion of treatment and financing capabilities that effectively doubles market size in this scenario. However, for this and other scenarios, financial prospects for drugs are poor when market penetration is assumed to be at levels comparable to those of LAAM and naltrexone.
The "Guaranteed Handoff" scenario considers the decision facing a company when the government is offering the rights to a drug that is well along in development in exchange for the company's finishing the development process and securing marketing approval. In addition, the government would (a) award orphan drug (or similar) status, (b) provide additional years of market protection from generics, and (c) guarantee purchases for up to 125,000 daily users for the years in which market protection, i.e., (a) and (b), apply. In this scenario, the risk-reward tradeoff is improved by effectively decreasing a company's investment and shortening the time to product launch. Even so, this scenario indicates that some combination of other incentives, such as extended orphan-like protection and a wider or more assured market in the form of guaranteed purchases of a set volume of the drug at an attractive price, may be required to make the arrangement sufficiently attractive to a company. The PAR and NPV generated by such a scenario may be more in line with the thresholds of smaller companies rather than larger ones.
The "Vaccine" scenario poses more of an outlier set of market conditions involving a promising medication that could be taken just once a year (e.g., vaccine with annual boosters). As in the "Guaranteed Handoff" scenario, this involves initial government development of the medication and an offer early in development to transfer rights to a company to bring the product to market. In this scenario, aside from extended generic protection, the government provides for a substantial, assured market in the form of guaranteed purchases at a premium price for a number of users that is twice the current number of daily enrollees in treatment for cocaine abuse. This scenario helps to illustrate that extraordinary conditions may be required to bring PAR and NPV over the thresholds sought by the larger pharmaceutical companies.
The "Second Indication" scenario portrays a decision about whether to pursue a cocaine abuse indication for a drug if doing so might jeopardize a currently successful market for the drug for another indication. Here, it is assumed that the additional development costs required to secure approval for the second indication would be relatively small, and that orphan-like status could be secured for the cocaine abuse indication. Under a base scenario with a moderate price and 50 percent market penetration, the drug would yield a positive NPV and a modest PAR that falls below large company standards but that might be more palatable to smaller companies. Higher prices could push the PAR over the higher thresholds, but such prices would exceed those for LAAM and naltrexone. This scenario illustrates how conservatism regarding expectations for price and market penetration alone can stall a project. Aversion to the prospects of substance abuse stigma transferring to an already successful product may be secondary, but it could contribute to outweighing any perceived financial returns of a second indication strategy.
Case Studies
The purpose of the case studies is to gain insight into the experiences of companies that are relevant to developing and marketing medications for drug abuse and addiction. These include two in-depth case studies of LAAM for heroin addiction and naltrexone for heroin addiction (as Trexan) and alcohol addiction (as ReVia), as well as two smaller case studies on clozapine for schizophrenia and Nicorette for smoking addiction.
The four case studies have several elements in common, particularly with regard to certain aspects of their target patient populations. Three of the four drugs involve treatment for substance abuse, including LAAM and naltrexone for heroin addiction, naltrexone for alcoholism, and Nicorette for smoking. Clozapine was included in this study because the market for schizophrenia pharmacotherapies was regarded as sharing certain characteristics with the market for substance abuse pharmacotherapies including: 1) relatively small market size, 2) treatment funding primarily through public sources, 3) and some patients who need help caring for themselves and complying with medication.
Each of the case studies provided important market lessons. The experience with LAAM demonstrates that the existing delivery system poses significant market barriers due to, e.g., state-by-state rescheduling processes, the methadone orientation of clinics, higher price relative to methadone, limited clinic budgets, and staff resistance to change. Naltrexone demonstrates the importance of understanding factors that affect patient compliance, notwithstanding its excellent pharmacological properties. In this instance, most patients preferred methadone to naltrexone, a non-addictive medication. Further, without being properly linked to sufficient and appropriate psychosocial therapy, a pharmacologically effective medication may not be successful in treating substance abuse. Naltrexone has been unable to gain acceptance into alcoholism treatment given resistance by providers and payers.
The experience with clozapine shows that a high cost of treatment (due in part to required adjunct treatment involving weekly patient monitoring for possible serious side effects) severely limited market penetration. For Nicorette, which entered the market as a prescription drug, the minimal distribution barriers and approval for over-the-counter use boosted sales and led to development and introduction of multiple competing products.
The government played a key role in the development of three of the four case study drugs by lowering some of the market barriers, particularly by funding development work, including clinical trials. For three of four of these case study drugs, the federal government funded a significant portion of the pre-clinical and clinical research necessary for FDA approval. As a group, the four case studies provided examples of other favorable government interventions, including FDA fast-track approval (LAAM, clozapine, and Nicorette) and modified phase IV clinical trial requirements (ReVia), market exclusivity (orphan drug status or other market protection for all four drugs), and mandated Medicaid coverage (clozapine).
Critical Market Barriers
Many of the market barriers identified in the 1995 IOM report were confirmed through the sources used for this study. Although no new general types of new market barriers were identified in this study, certain ones were elaborated or described in a more contemporary context.
Two main categories of market barriers emerged from this study. Critical barriers are those that must be lowered or eliminated in order for pharmaceutical firms to regard the prospects for developing cocaine addiction medications as financially feasible. Non-critical market barriers are those that, if lowered or eliminated, may enhance, though perhaps only marginally, the financial outlook for developing cocaine addiction medications only if the critical barriers are also lowered. That is, without movement on the critical barriers, lowering non-critical ones would be unlikely to transform an otherwise unattractive market into an attractive one.
Among the diverse market barriers perceived by the industry, three emerged as critical in this study, i.e., those that would have to be lowered or eliminated in order to begin to make new drug development attractive to pharmaceutical companies:
- Small and uncertain market for cocaine addiction and abuse pharmacotherapy;
- A substance abuse treatment system that limits access to this market; and
- Limited and uncertain payment for pharmacotherapy for this indication.
Critical Barrier 1: Small and Uncertain Market for Cocaine Addiction and Abuse Pharmacotherapy
The small size and uncertainty of the market for cocaine pharmacotherapies constitutes a critical barrier to development of a cocaine abuse pharmacotherapy. Although all of the company executives interviewed for this study agreed that the total number of cocaine users is appreciable, they recognized that the feasible market for a cocaine abuse treatment is likely to be much smaller than the absolute number of people that use cocaine. Representatives of one pharmaceutical company use a conservative estimate of the number of heavy cocaine users that is about half of the level of 2 million cited in this report.
Uncertain market penetration was another reason for the skepticism in industry. Interviewees stressed that potential patient compliance problems and limited access to patients made them uncertain about the true market size for cocaine treatment. Representatives of two companies noted that most publicly-funded treatment centers are managed by non-physicians who tend to oppose the use of drugs to treat substance abuse, which such staff regard as a "behavioral" condition, thereby further restricting the potential sale of these drugs.
Critical Barrier 2: A Substance Abuse Treatment System that Limits Access to the Market
There are multiple, interrelated aspects of the current substance abuse treatment system that limit the market prospects for any new pharmacotherapy for cocaine addiction. These limitations are apparent in the case studies, were raised by company executives interviewed for this study, and are corroborated by modeling of certain scenarios. Sales of LAAM and naltrexone were restricted by the limited number of heroin and alcohol treatment programs and the limited capacity of these programs. Whereas 25 percent of opiate addicts receive treatment from the methadone maintenance programs, only about 5 percent of those afflicted by alcohol abuse and dependence are in alcohol treatment centers. Distribution of LAAM is restricted to maintenance programs as required by The Narcotic Addict Treatment Act of 1974. Prescription of naltrexone is recommended to be linked to enrollment in comprehensive treatment centers in order to improve patient outcomes. In contrast, because Nicorette, originally a prescription medication, is now an over-the-counter formulation, patients need not visit a treatment center or a provider to obtain treatment, vastly expanding the drug's potential market.
The lack of medical treatment models in substance abuse treatment centers contributes to their being a critical market barrier. Pharmaceutical company executives cited an "anti-medication" climate among the publicly-funded treatment center staff that would severely limit sales of pharmacotherapies through treatment centers. Interviewees indicated that the large number of non-physicians (sometimes referred to as "non-prescribers") at treatment centers often have strong anti-medication sentiments. As noted above, recent surveys that have examined staffing patterns confirm that the substance abuse treatment system involves little or no physician time in the treatment of patients. This observation was confirmed in the LAAM and naltrexone (Trexan) case studies, which found that treatment decisions and funding for heroin addiction are often mediated by state-level substance abuse program administrators who often do not have clinical backgrounds.
Critical Barrier 3: Limited and Uncertain Payment for Pharmacotherapy
Industry decision makers recognize the heavy reliance of the substance abuse market on federal, state, and local government reimbursement. The perception among the drug companies is that many cocaine addicts do not have private insurance and rely on federal and state government sources for treatment, and that only a portion of those individuals with private insurance use their benefits for drug abuse treatment. One executive noted that substance abuse services continue to be subsumed under mental health benefits of entitlement programs, and that the overall budget for mental health services continues to shrink in light of other competing health priorities.
Payment status is a recognized barrier for LAAM, naltrexone, and clozapine. Treatment for heroin addiction (e.g., LAAM and naltrexone) has been funded primarily through federal and state budgets, making reimbursement difficult for pharmaceutical companies. As noted above, price sensitivity to a cocaine medication is another aspect of payment that poses a critical market barrier because price resistance may limit market size.
Industry Perception of Science Base Readiness
There was a divergence of opinion among the pharmaceutical company interviewees about the readiness of the science base for cocaine pharmacotherapies. Representatives of two companies expressed skepticism about the readiness of the science base. One representative indicated that current limitations stem from a lack of understanding regarding the biological and genetic basis of addiction. A representative of a different company indicated that the current science base for achieving long-term efficacy for cocaine abuse and addiction is very weak. Furthermore, scientists from one company judged that the probability of a scientific breakthrough in the area of cocaine abuse and addiction in the near future is very low. In contrast, representatives of another pharmaceutical company indicated strongly that the science base is ready, and consequently that it is no longer a market barrier to development of cocaine pharmacotherapies. This company also reported that it had successfully identified several drug candidates that exhibited cocaine blocking activities in both in vivo and in vitro models. The extent of company interviews was limited by the scope of this project.
Overcoming Critical Market Barriers
Any public policies intended to improve opportunities for developing pharmacotherapies for cocaine addiction must address the three critical barriers described here. It is not within the scope of this study to identify or analyze specific public policies to promote development or marketing of pharmacotherapies for substance abuse. Nevertheless, during the course of this study, certain types of strategies or initiatives emerged that would serve to lower these barriers and make the development of new pharmacotherapies for cocaine abuse more attractive to the pharmaceutical industry, as follows:
- Government funding of a considerable portion of new drug development;
- Expansion and enhancement of the substance abuse treatment system;
- Guaranteed market (e.g., purchase orders for minimum volumes of a medication); and
- Extended market exclusivity (e.g., orphan drug or similar status).
The pertinence of such actions is supported by lessons from the case studies, suggestions raised by interviewees, and results of modeling diverse scenarios of new pharmacotherapy development described in this report. These strategies are consistent with certain of the strategies recommended elsewhere, e.g., certain ones raised by the IOM (1995), and merit further attention.
Introduction
This report provides an analysis of the market barriers to the development of pharmacotherapies for cocaine abuse and addiction. The topic of drug addiction continues to raise significant medical, social, and economic public health concerns, and pharmacotherapy offers a means for improving the treatment of drug addiction. Only a handful of medications have been developed and received FDA approval for treatment of opiate addiction during the past 30 years, but these have had limited success and better medications are needed. Further, no pharmacotherapies have been approved for the treatment of cocaine addiction, and, according to a 1995 Institute of Medicine (IOM) report on the development of anti-opiate and anti-cocaine medications, there has been a reluctance on the part of the pharmaceutical industry to enter the field of anti-addiction products because of considerable market disincentives.
The 1995 IOM report found that the major disincentives to pharmaceutical R&D for anti-addiction medications include an inadequate science base on addiction and the prevention of relapse, and an uncertain market environment regarding e.g., treatment financing, lack of trained specialists for treatment of drug addiction, federal and state regulations, market size, pricing issues, societal stigma, liability issues, and difficulties in conducting clinical research.
The purpose of this report is to review and analyze market barriers (both real and perceived) to the development of pharmacotherapies for the treatment of cocaine abuse and addiction. This report provides the Department with information and analyses in the following four areas:
- characteristics of the market for cocaine abuse and addiction pharmacotherapies
- real and perceived market barriers to cocaine pharmacotherapy development
- case studies of the development and marketing of selected pharmacotherapies
- private industry's perception of the readiness of the science base for developing such medications
This report is not intended to analyze or make recommendations concerning any particular government initiatives or other policies that may be under consideration by the Department regarding treatments for cocaine abuse.
Pursuant to the purpose of this project, ASPE contracted with The Lewin Group to:
- conduct a market analysis for a prospective cocaine medication;
- conduct case studies of selected pharmacotherapies (LAAM, naltrexone, clozapine, and Nicorette) through literature reviews and interviews with key individuals involved in the development and marketing of these products;
- conduct interviews with representatives of several private firms (e.g., pharmaceutical companies, venture capital firms) to determine industry's views on market barriers (including the readiness of the science base for a pharmacotherapy for cocaine abuse and addiction).
The present document is organized as follows. First, it provides a market analysis from the perspective of a private firm considering an investment in the development of a cocaine pharmacotherapy. This includes discussions of the relationships among such factors as market size, price, and revenues as they relate to corporate decision making. Second, the report describes five hypothetical scenarios of company decision making regarding investing in new drug development for treating cocaine addiction. These scenarios help to illustrate how various market factors and financial parameters may affect drug development decisions. Third, the report documents the case experiences of pharmaceutical firms that have developed pharmacotherapies for substance abuse and addiction, e.g., LAAM and naltrexone. Also provided are case study reports of two non-substance abuse drugs, clozapine and Nicorette, in order to draw lessons from other disease areas that may be applicable to future development in the cocaine area. Finally, the report addresses market barriers to the development of cocaine pharmacotherapies, including readiness of science base, as described by scientific, marketing, and other executive-level representatives of several key private sector stakeholders (e.g., pharmaceutical companies, venture capital firms).
Study Methods
We used several sources to collect information about substance abuse pharmacotherapies (e.g., cocaine) and the market barriers to the development of such drugs. We completed a market analysis for cocaine pharmacotherapy; several hypothetical scenarios of pharmaceutical decisionmaking; four case studies of selected pharmacotherapies; and detailed interviews with five private firms (e.g., pharmaceutical companies, venture capital firms) that have real or potential interest in substance abuse and/or other CNS drug development. The following sections briefly describe our general study methods according to the various study modules; more detailed study methods are described in the appendices.
Market Analysis for a Prospective Cocaine Medication
We performed a market analysis for a prospective cocaine medication from a pharmaceutical company's perspective. The purpose of this market analysis was to estimate how costs and revenues would be accrued over time in the development and commercialization of a prospective cocaine medication.
We first focused on estimating the market size for substance abuse treatment, and, more specifically, for cocaine treatment. Also, we described both the revenue potential and potential sources of payment for cocaine treatment.
As such, we compiled data on the following:
- the prevalence and incidence of opiate and cocaine addiction (including available demographic information such as age, sex, and income of addicted individuals)
- current rates of treatment for opiate and cocaine dependence
- existing patterns of service delivery and financing for drug treatment
We relied primarily on data in the public domain. Data sources, grouped according to the type of information contained in the data set, are shown in Figure 1 (below).
| Type of Information | Data Sources Utilized |
|---|---|
| Need for Treatment |
Abt Associates Report: Synthetic Estimation Applied to Prevalence of Drug Use |
| Service Utilization and Financing |
- Substance Abuse - Mental Health |
In addition, we conducted a limited but targeted literature search of the gray literature (e.g., MEDLARS, Dialog, National Newspaper Index, other published studies and commissioned reports, and product packaging and marketing materials) to look for existing market size estimates of the anti-depressant drug market that we could compare to our opiate and cocaine addiction market size estimates.
Market Analysis Model
Drug development decisions of pharmaceutical companies are primarily based on business and economic factors--essentially whether a particular potential medication might be sufficiently profitable if it is successfully developed. Expected profitability of a particular product is determined by a combination of such factors as:
- development cost
- sales of the product
- cost of manufacturing, distributing and marketing the product
- duration of the development and product/sales life.
In order to analyze and better explain the development decisions of pharmaceutical companies, we developed a market analysis model that incorporates these factors. This model allows simulation of the economics driving a particular development decision and assessment of the sensitivity to various market and policy scenarios. The potential commercial attractiveness of a cocaine medication is assessed using this model, and selected values are believed to be plausible. In addition, the model is used to examine the sensitivity of conclusions to variation in particular factors The model was built on a spreadsheet, and has a user-friendly interface to enable determining how modification of inputs affects the two main output variables, PAR and NPV. Details about the model are given in the appendix.
For purposes of designing this model, it was assumed that a pharmaceutical company's assessment of the attractiveness of a potential medication/product can be based largely on certain financial indicators, e.g., product net present value (NPV) and peak annual revenue (PAR) of the product. Using information from other aspects of the project, various potential market conditions were translated into financial and other parameters that were fed into the model to generate NPV and PAR calculations. The input variables used in the model are shown in Figure 2.
| Uncapitalized R&D costs | Orphan drug (or similar) status |
|---|---|
| Stage of entry | Years of orphan drug extension |
| Discount rate | Orphan drug (or similar) tax advantage |
| Wholesale price | Years post-launch to competing drug |
| Peak market size | Years to replacement by competing drug |
| Weeks of prescription | First year MMDA* costs |
| Expected peak prescriptions | Duration of marketing campaign |
| Years post-launch to peak prescriptions | MMDA costs during marketing campaign |
| Years to patent expiration | MMDA costs after marketing campaign |
| *MMDA: manufacturing, marketing, distribution, administration | |
A listing of terminology relevant to this report is provided below in Figure 3.
| Figure 3: Definitions of Selected Terms for Market Analysis |
|---|
|
Capitalized cost: The value of research and development expenditures plus the accrued interest costs on those outlays. Discount rate: The interest rate used to convert future cash flows to their present value. Expected return: Average of possible returns weighted by their probabilities. Net present value (NPV): The difference between the present value of all cash inflows from a project and the present value of all cash outflows required for the investment, using the required rate of return to calculate present values. In general, investments are accepted that have positive NPVs. Opportunity cost of capital: The expected return that is forgone by investing in a project rather than in comparable financial securities. Also known as hurdle rate or cost of capital. Peak Annual Revenue (PAR): The highest annual revenue achieved by a product during its market life. Present value: Discounted value of future cash flows Required rate of return (RRR): The minimum acceptable rate of return on an investment. In general, investments are accepted that offer rates of return in excess of their discount rate or costs of capital. Return on investment (ROI): Income divided by investment. |
|
Sources: Brealey RA, Myers SC. Principles of Corporate Finance. Fourth Edition. New York: McGraw-Hill, 1991. Horngren CT, Foster G. Cost Accounting. A Managerial Emphasis. Seventh Edition. Englewood Cliffs, N.J.: Prentice Hall, 1991. U.S. Congress. Office of Technology Assessment. Pharmaceutical R&D: Costs, Risks and Rewards. Washington, DC: U.S. Government Printing Office, 1993. |
Scenarios of Company Decision Making
The Lewin Group adapted the quantitative model to test hypothetical scenarios of company decision making. Modeling of the scenarios made it possible to examine whether a particular product development opportunity might appear more or less attractive to pharmaceutical companies and their investors. The scenarios help to illustrate some of the key barriers and other limitations to development of medications for cocaine abuse, as well as the extent to which certain types of financial and policy options might reduce such barriers.
The scenarios are entirely hypothetical works drafted by The Lewin Group. Aspects of the scenarios were drawn from the literature on drug development as well as information gleaned from the market analysis, case studies, interviews with private firms, and other sources used for this study. The scenarios do not represent any suggestion or intent by the government to adopt any policies described in the scenarios. Certain policy options posed in the scenarios are already in existence, e.g., orphan drug or similar status; others have been discussed in the 1995 IOM report referenced in this study.
Case Studies
For each of the case studies, we used several different kinds of sources to collect information on the market barriers to the development of each drug. For each case study, we conducted a thorough literature search, including peer-reviewed sources, and the gray literature. The information gathered from print materials was supplemented with interviews of key individuals from industry and government who were involved in the development, evaluation, and commercialization of the product.
The literature review was conducted by a direct search of MEDLARS databases, including MEDLINE (citations of peer-reviewed journal literature), HealthSTAR (citations of journal literature and other sources in health services research, technology assessment, and health planning), and HSRProj (citations of recent and ongoing health services research funded by government and the private sector). Among the bibliographic databases, we focused in particular on MEDLINE 1966 - present and HealthSTAR 1984 - present to obtain information on clinical trials, product development and marketing. It was critical to examine articles that dated back to the 1960's for all of the case studies, as some of the early preclinical and clinical development happened well over thirty years ago. The search was restricted to English-language publications particularly because we were focusing on issues related to market barriers in the US. In addition, a similar search was conducted in Dialog databases which compiles journal articles from a variety of database sources including Scrips, ABI/INFORM ®, FDA Pink Sheet, and IAC Trade and Industry Database. The MeSH (Medical Subject Headings) terms used to identify articles for the LAAM, naltrexone, clozapine, and Nicorette case studies are shown in Appendix C.
To identify articles pertinent to the development of LAAM, the search strategy included both LAAM and methadone, a medication currently used by all opioid treatment programs for maintenance therapy. The use of methadone in the search facilitated the discovery of articles that compared the two medications.
For naltrexone, the search strategy included both NALTREXONE, the generic name of the product, and TREXAN, the trade-name of the product for the opioid indication. The use of TREXAN as part of the search strategy effectively reduced the scope of the literature search to the key opioid indication, as many disparate research projects were published using naltrexone, unsuccessfully to treat a variety of other conditions. Since naltrexone is currently indicated for the treatment of two conditions (opioid addiction and alcoholism), our literature review encompassed the key issues to the development of naltrexone for both indications.
For the other two case studies, MeSH terms for CLOZAPINE and SCHIZOPHRENIA were used to identify articles on clozapine, and MeSH terms for Nicorette and smoking cessation therapy were used to identify articles on Nicorette. Nicotine polacrilex (the generic name for Nicorette) was also used in our search strategy for Nicorette, and proved useful in gathering articles on the compound's marketing.
Interviews with Private Firms
We interviewed five private firms, one at a time, to explore and characterize their views on market barriers to the development of cocaine abuse pharmacotherapies and on the readiness of the science base in developing such medications. One of the companies was a large pharmaceutical company; one was a large pharmaceutical company with a strong CNS focus; one was a small biotechnology company with a substance abuse pharmacotherapy in its portfolio; and two were venture capital firms that had investments in health care and biotechnology.
Information gathered from these interviews appears in the Market Barriers section of the report. Appendix E includes a list of selection criteria of the private firms interviewed and a detailed discussion guide that served as our interview protocol.
The next section of the report presents the main findings from our study. Our findings are organized along the following dimensions:
- Market analysis for a prospective cocaine medication
- Scenarios of company decisionmaking
- Case studies of selected pharmacotherapies
- Market barriers to cocaine pharmacotherapy development.
Market Analysis for a Prospective Cocaine Medication
The market analysis estimates how costs and revenues accrue over time in the development and commercialization of a prospective cocaine medication, and presents plausible scenarios of prospective pharmaceutical companies and their drug development decisionmaking process. For pharmaceutical companies, the risk associated with developing and marketing a cocaine medication is considerable. While it is possible to construct plausible scenarios in which half a million patients per day would be using a cocaine medication, a user population that is half or one quarter this size is more plausible. Notwithstanding recent advances in the science base for cocaine addiction pharmacotherapies, it is unrealistic to expect most pharmaceutical firms to consider investing in such products in the near future.
In general, pharmaceutical firms seek competitive returns on their investment. As noted above, one threshold used in the industry is the net present value (NPV) of the product. Another financial threshold used by pharmaceutical firms is a drug's expected peak annual revenue (PAR). A conservative threshold target PAR for a prospective new product in the pharmaceutical industry is about $200 million for a single drug, although the larger companies tend to seek PARs of $250 to 500 million or more. This report uses a market model to analyze the NPV using plausible estimates of relevant parameters, including market size, market penetration, costs of developing a medication, and price. This model is presented and discussed later in this document.
This section provides national estimates of drug abuse in general and for cocaine abuse in particular; national expenditures for alcohol and drug abuse, and for cocaine abuse and addiction; payment sources for cocaine treatment; and a scenario involving potential revenues for a potential pharmacotherapy for cocaine abuse and addiction.
Need for Drug Treatment in the U.S.
Figure 4: Summary of Estimates of Need for Drug Abuse Treatment in U.S
Several studies conducted since 1990 have estimated the need for drug abuse treatment in the U.S. These estimates provide points of reference for the overall magnitude of potential markets for drug abuse medications.
The primary findings of these studies are summarized:
- The overall magnitude of the population in need of drug abuse treatment (for cocaine, heroin, amphetamines, marijuana, etc.) is approximately 3.5 to 4.5 million persons. There is another population of similar magnitude with a lower or marginal potential to benefit from treatment. (See estimates for Level 1 and Level 2 need by Woodward et al., in press).
- The number of opiate addicts (primarily heroin users) is at least 500,000 persons (Rhodes et al. 1993; Rhodes et al. 1995; Hamill and Cooley 1990). This estimate is similar to estimates of this population dating from the early 1970s (see Appendix D).
- The magnitude of the population in need of cocaine treatment is at least 2 million persons, based on several similar estimates (see Appendix D, Rhodes et al. 1993; Rhodes et al. 1995; Everingham and Rydell 1994; SAMHSA Office of Applied Studies 1997; U.S. Senate Judiciary Committee 1990).
Prevailing national estimates for the need for treatment of substance abuse, in general, and cocaine abuse, in particular, are based on results of the National Household Survey on Drug Abuse (NHSDA), sponsored by the Substance Abuse and Mental Health Services Administration (SAMHSA; Office of Applied Studies, 1996) and are summarized in Figure 4 (above). Due to insufficient coverage of selected populations, e.g., criminal justice populations, NHSDA data likely provide low estimates of treatment need (Gerstein and Harwood, 1990). In order to generate more realistic estimates of need for treatment, most analyses augment the estimates from the NHSDA with data from other sources. Such estimates are used as well to gauge how well the national treatment system is able to deliver care to the population in need, and to estimate the need for additional funding for substance abuse treatment (Figure 5 below).
| Source | |
|---|---|
| NHSDA POPULATION ESTIMATES (OAS 1996) | |
| Any illicit drug use in past month (1995) | 12,800,000 |
| SAMHSA (Woodward et al., in press) | |
| Total | 7,100,000 |
| Level 1 Need (1994) | 3,500,000 |
| Level 2 Need (1994) | 3,600,000 |
| IOM METHODOLOGY (Gerstein & Harwood, 1990) | |
| (Harwood et al., 1993) Total Estimate of Treatment Need (1991) | 4,887,000 |
| Total Estimate of Treatment Need (1987-1988) | 5,455,000 |
| Household Population: Clear need (1987-1988) | 1,500,000 |
| Household Population: Probable need (1987-1988) | 3,100,000 |
| Homeless(sheltered, street, and transient) | 170,000 |
| Correctional Custody | 320,000 |
| Probation and parole | 730,000 |
| Pregnancies (live births) | 105,000 |
| Less overlaps | (470,000) |
| EPIDEMIOLOGIC CATCHMENT AREA (Regier et al., 1993)1 | |
| Any Drug Disorder (1980 data, applied to 1990 pop.) | 5,742,000 |
| NATIONAL COMORBIDITY SURVEY (Kessler et al., 1995) | |
| Prior 12-month drug-dependence plus abuse (1992) | 4,663,000 |
| 1 Based on 1980 data with a 16.5% adjustment to account for the population increase from 1980 to 1990. This is an underestimate of cocaine abuse, as cocaine use increased dramatically in the late 1980s. | |
In the U.S., a specialized treatment system delivers the vast majority of substance abuse treatment services, including treatment for cocaine and opiate addiction (Harwood et al., 1994; Rice et al., 1990; Harwood et al., 1984). The great bulk of substance abuse treatment services are delivered by institutional providers; only small shares of services are delivered by physicians in private practice (including general practitioners and psychiatrists), private practice mental health specialists, and general hospitals.
Based on findings of the 1993 National Drug, Alcohol Treatment Unit Survey (or NDATUS, Office of Applied Studies, 1995), the treatment system includes 11,500 institutional providers, including specialized freestanding clinics, mental health clinics, and specialized hospitals and hospitals with specialized units. Among these 11,500 institutional providers, the overall one-day census, i.e., people currently enrolled in substance abuse treatment, was 944,000 patients (Figure 6). NDATUS estimates that 38,000 of these persons are in the criminal justice population. Setting aside this likely unreliable estimate, there are approximately 900,000 patients currently enrolled in substance abuse treatment.
| Number of Providers, and Daily Treatment Enrollment | ||||
|---|---|---|---|---|
| Institutional Setting | ||||
| TOTAL | 11,496 | 121.1 | 823.1 | 944.2 |
| Free-Standing Non-residential | 5,038 | 1.7 | 502.0 | 503.7 |
| Community Mental Health Center | 1,413 | 4.2 | 136.4 | 140.7 |
| General Hospital (incl. VA) | 1,267 | 14.0 | 81.8 | 95.8 |
| Residential Facilities | 499 | 47.0 | 23.1 | 70.4 |
| Specialized Hospitals | 955 | 8.7 | 14.0 | 22.7 |
| Halfway House/Recovery Home | 1,530 | 18.9 | 5.4 | 24.3 |
| Correctional Facilities | 313 | 18.4 | 20.0 | 38.4 |
| Other Types | 481 | 7.9 | 40.3 | 48.2 |
| Source: NDATUS, Office of Applied Studies, SAMHSA, 1995. | ||||
The total number of people that receive substance abuse treatment services at least once during a year is not directly available from NDATUS because, although all institutional providers provided data for current enrollees, some did not provide data for the total number of patients served during the year. Among the institutional providers that did report on this, 2.8 million patients were served at least once during 1993. Among this same set of institutional providers the overall one-day census was 720,000. This yields a turnover in daily census of about 3.9. However, that is an overestimate of turnover, because some patients enroll with more than one institutional provider during any given year. Based on findings from The California Drug and Alcohol Treatment Assessment (CALDATA; Gerstein et al., 1994), Denmead et al. (1995) estimated the true turnover rate to be about 3.6. Applying this turnover rate to the daily census of 900,000 from NDATUS yields 3.2 million people that were treated for substance abuse at least once during 1993.
Estimate of Cocaine Users in the U.S.
The following paragraph provides a summary of some plausible estimates of cocaine use in the United States. The remainder of this section describes the national surveys on drug abuse from which the estimates of cocaine use are derived.
There are in excess of 2 million addicted or "heavy" cocaine users in the U.S. Of these, about 800,000 to 900,000 enter treatment at least once in a given year. On any given day, there about 250,000 cocaine users enrolled in treatment (i.e., at a residential facility or have been served at an ambulatory treatment center within the previous 30 days). Of these 250,000 currently enrolled users, roughly 150,000 are primary cocaine abusers and 100,000 are secondary cocaine abusers. Thus, of all heavy cocaine users, slightly more than 10 percent are enrolled in treatment on any given day.
While there is no definitive method for estimating the number of cocaine abusers in treatment on any given day, estimates of cocaine users currently enrolled in treatment can best be derived by applying estimates of the proportion of all substance abusers who are cocaine abusers to the NDATUS estimate of 900,000 substance abusers.
Several recent surveys have estimated that up to 17 percent of patients entering specialty substance abuse treatment have primary cocaine abuse (e.g., smoked or other) (Figure 7 below). The State Alcohol and Drug Abuse Profile (SADAP) surveys suggest that the proportion of primary cocaine admits has increased during the 1990s; however, it contains no information on cocaine abuse secondary to other substances. Application of this 17 percent rate yields an estimate of 150,000 daily patients with primary cocaine abuse.
| Study (Year of Data) | ||||||
|---|---|---|---|---|---|---|
| TEDS, 1995 | 1.328 admits | 17.1 | 19 | 8.3 | 10.7 | 36.1 |
| SADAP, 1994 | 1.826 admits | 17.9 | - | - | - | - |
| SADAP, 1993 | 1.755 admits | 15.7 | - | - | - | - |
| SADAP, 1992 | 1.793 admits | 14.8 | - | - | - | - |
| SADAP, 1991 | 1.976 admits | 11.4 | - | - | - | - |
| SADAP, 1990 | 1.910 admits | 11.4 | - | - | - | - |
| DSRS I, 1990 | 0.720 clients | 10.7 | - | - | "with" 18.9 | 29.6 |
| DSRS II, 1990 | 1.048 dischs | - | - | - | - | 38.5 |
Sources: TEDS: Office of Applied Studies, SAMHSA, 1997. SADAP (all): National Association of State Alcohol & Drug Abuse Directors, various. DSRS Phase I: Brandeis University, 1993a. DSRS Phase II: Brandeis University, 1993b. | ||||||
The Treatment Episode Data Set 1992-1995 (TEDS) survey indicates that an additional 19 percent of substance abuse enrollees are secondary cocaine abusers, i.e., are primarily abusing one or more other substances such as alcohol or heroin. This yields an estimated 170,000 secondary cocaine abusers. However, because cocaine abuse may be the third or fourth drug problem for many of these patients, a more conservative estimate of 100,000 is used. Adding this conservative estimate of 100,000 patients to the SADAP estimate for patients with primary cocaine abuse (150,000) yields a combined estimate of 250,000 primary or secondary cocaine abusers.
The SADAP and TEDS surveys must be used advisedly. First, they draw data exclusively from publicly-reimbursed specialty providers. Second, although such providers have 80 percent of the daily patients (among providers reporting funding sources), there is some uncertainty about how complete and representative these surveys are in their coverage of treatment admissions. TEDS reports on 1.3 million admissions and SADAP on 1.9 million admissions, both of which are purported to be a census of admissions to their provider frames. Other studies have estimated that there are over 3 million admissions to specialty treatment providers per year (Harwood et al., 1994; Denmead et al., 1995; Office of Applied Studies, 1997). Because SADAP and TEDS cover publicly-reimbursed patients only, they may over-represent cocaine abuse relative to the entire population, which may have somewhat lower levels of cocaine abuse relative to all types of drug abuse.
The estimate of the number of cocaine addicts varies somewhat. The Drug Services Research Surveys (DSRS) indicate that by 1990 there were in excess of 200,000 cocaine patients per day (Brandeis University, 1993a, 1993b). In linked studies, DSRS examined drug problems of patients according to the one-day census of treatment (Phase I) and annual discharges from treatment (Phase II). Based on Phase I findings, about 30 percent (i.e., 210,000) of the 720,000 patients enrolled with "drug" or "drug and alcohol" treatment providers (excluding "primary alcohol" providers) had a diagnosable cocaine problem. Although little detail was provided for the estimates, it can be deduced that cocaine was the unambiguous primary problem for about 11 percent (75,000) of all enrollees and that cocaine was accompanied by, and may or may not have been secondary to, alcohol for the other 135,000 enrollees.
In Phase II of DSRS, data on type of substance abuse were available for only 81 percent of the 1.05 million discharged patients. Of these patients, 38.5 percent reported having drug or combined drug and alcohol abuse. Application of proportion to the NDATUS estimate of 620,000 current enrollees in drug or combined drug and alcohol treatment (i.e., 900,000 total substance abusers minus 280,000 alcohol-only abusers; Office of Applied Studies, 1995) yields an estimate of about 240,000 cocaine patients per day.
The estimate of a total of 900,000 cocaine users who are treated at least once in a given year is derived by applying the turnover rate of 3.6 (noted above) to the 250,000 persons with primary or secondary cocaine diagnoses. Figure 8 (below) provides a summary of the general characteristics for cocaine treatment in the U.S.
| Measure of Cocaine Treatment Market | Estimate |
|---|---|
| Need for cocaine treatment | ³ 2 million heavy users |
| Enrollment in treatment (primary cocaine diagnoses plus 60% of secondary diagnoses) | 250,000 patients per day |
| Specialty providers treating cocaine abusers | 11,500 providers |
| Annual treatment episodes (primary cocaine diagnoses) | 800,000 to 1.0 million admissions per year |
| Individual cocaine abusers treated per year | 900,000 persons |
| Spending on treatment | $2.1 billion |
| Spending on treatment per cocaine patient | $23.00 per day enrolled $9.00 per day in outpatient |
| Source: Analysis by The Lewin Group. | |
National Expenditures on Cocaine Treatment
National expenditures for specialized alcohol and drug treatment were about $6.5 billion in 1993, or $19.71 per-patient-per-day for all treatment (Figure 9 below). Approximately $2.6 billion of this amount was spent on outpatient care (i.e., $1.94 billion and $0.65 billion for outpatient-standard and outpatient-intensive care, respectively), or only about $8.89 per-patient-per-day for outpatient care. In contrast, expenditures for the 103,000 daily enrolled patients receiving 24-hour care (i.e., all modalities except for detox-ambulatory and both outpatient modalities) were $3.87 billion, or about $103 per-patient-per-day. Expenditures for the approximately 80 percent of substance abuse patients who are in standard outpatient care were $7.38 per-patient-per-day.
| MODALITY OF CARE | Daily Clients Enrolled | Annual Revenue ($ in millions) | Revenue per Patient ($ per day) |
|---|---|---|---|
| Detox-Ambulatory | 6,700 | $72 | $29.66 |
| Detox-Freestanding | 6,400 | $267 | $114.33 |
| Detox-Hospital | 7,300 | $1,005 | $375.22 |
| Rehab-Hospital | 14,300 | $881 | $168.53 |
| Rehab-Short Term | 15,000 | $587 | $107.53 |
| Rehab-Long Term | 60,100 | $1,127 | $51.42 |
| Outpatient-Intensive | 78,000 | $649 | $22.77 |
| Outpatient-Standard | 719,100 | $1,937 | $7.38 |
| TOTAL | 906,800 | $6,525 | $19.71 |
| Source: Office of Applied Studies, SAMSHA, 1995 | |||
The estimates of revenue were based on the NDATUS compilation of $3.9 billion from centers that reported revenue, plus an estimated $2.6 billion to account for centers that did not report revenue. Estimates were based on the number of daily patients, by type/modality of care, multiplied by revenue per day (column 4 of the figure above) from reporting "single modality" providers.
Expenditures for specialty treatment for cocaine abuse total approximately $2.1 billion, including about $1.26 billion for the approximately 150,000 primary cocaine addicts and $0.84 billion for the approximately 100,000 secondary cocaine addicts (Figure 10 below). Expenditures averaged $9.00 per day per patient enrolled in standard outpatient treatment and $23.00 per day per patient enrolled in any modality of care (i.e., including outpatient and inpatient care).
| All Primary Cocaine Addicts, Adjusted up by 2/3 for Secondary Addicts | ||||
|---|---|---|---|---|
| MODALITY OF CARE | Average Daily Enrollment | Annual Revenue ($ in millions) | Primary Cocaine Admits (percent) | Revenue for Primary and Secondary Patients ($ in millions) |
| Detox-Ambulatory | 6,700 | $72 | 2.2 | $3 |
| Detox-Freestanding | 6,400 | $267 | 17.2 | $77 |
| Detox-Hospital | 7,300 | $1,005 | 12.6 | $211 |
| Rehab-Hospital | 14,300 | $881 | 18.6 | $274 |
| Rehab-Short Term | 15,000 | $587 | 22.5 | $221 |
| Rehab-Long Term | 60,100 | $1,127 | 32.2 | $606 |
| Outpatient-Intensive | 78,000 | $649 | 22.4 | $243 |
| Outpatient-Standard | 719,100 | $1,937 | 14.5 | $469 |
| TOTAL | 906,800 | $6,525 | 17.1 | $2,103 |
| Source: Analysis by The Lewin Group. Daily enrollment and annual revenue (imputations for missing data) from 1993 NDATUS (OAS, 1996). Share of clients by modality from TEDS (OAS, 1997). Cocaine share by modality increased by 2/3 to adjust for share of secondary cocaine patients. | ||||
Although cocaine addicts account for 17.1 percent of all substance abuse admits, they account for higher percentages in the more intensive and expensive treatment settings such as rehabilitation centers and outpatient-intensive care (OAS 1997). Consistent with development of the estimate of daily cocaine addicts in treatment, the revenue estimate is based on 250,000 daily patients, including 150,000 daily primary cocaine patients and 100,000 daily secondary cocaine patients.
Sources of Payment for Cocaine Treatment
One of the major questions in developing a new cocaine medication is whether there will be sufficient and reliable payment for the medication. Payment for substance abuse treatment, including for cocaine abuse, remains heavily dependent upon government sources. Government provided almost 80 percent of the financing for substance abuse treatment in 1992 (Harwood et al., 1994). This represents only a modest shift to private sector sources since the 1982 NDATUS, which estimated that private sources (insurance plus out-of-pocket) accounted for about 6.5 percent of payments for specialty provider substance abuse treatment (Rice et al., 1990).
The limited sources with reliable data about payment sources in the substance abuse population suggest that only a fraction of patients seeking care for substance abuse treatment have either private insurance or sufficient earning capacity to reasonably afford a medication that would be taken for an extended period of time (e.g., several months or more). According to 1995 TEDS data (Figure 11 below), more than two-thirds of all enrolled cocaine abusers had no health coverage, and another 17.6 percent had Medicaid coverage. Only 9.1 percent of enrolled cocaine abusers had coverage through private insurance (e.g., Blue Cross/Blue Shield, HMO). Wages or salaries were the primary source of income for only 28.1 percent of enrolled cocaine abusers, while 51.9 percent were either on public assistance or had no income. More than three-fourths of the enrolled cocaine abusers were either unemployed or not in the labor force, and nearly 40 percent had not graduated from high school.
| Indicators of Ability to Pay for Treatment | |||
|---|---|---|---|
| Expected Source of Payment | |||
| Self-pay | 33.9 | 30.3 | 27.6 |
| Private Insurance | 8.7 | 5.1 | 5.4 |
| Medicaid | 14.3 | 20.1 | 27.9 |
| Medicare | 0.9 | 0.8 | 0.9 |
| Other Govt. | 24.0 | 22.9 | 22.7 |
| No Charge | 11.2 | 14.9 | 8.2 |
| Other | 7.1 | 5.8 | 7.2 |
| 100.0 | 100.0 | 100.0 | |
| Type of Health Insurance | |||
| None | 67.0 | 68.9 | 61.0 |
| Medicaid | 13.4 | 17.6 | 23.7 |
| Private Insurance | 7.4 | 4.2 | 3.9 |
| Blue Cross/Shield | 3.5 | 2.2 | 2.1 |
| HMO | 3.1 | 2.7 | 3.4 |
| Medicare | 1.6 | 1.1 | 1.8 |
| Other | 4.0 | 3.4 | 4.1 |
| 100.0 | 100.0 | 100.0 | |
| Primary Source of Income | |||
| Wages/salary | 38.5 | 28.1 | 22.3 |
| Public Assistance | 17.7 | 24.6 | 33.4 |
| Retirement/Pension | 0.9 | 0.3 | 0.3 |
| Disability | 4.5 | 3.5 | 3.3 |
| Other | 20.1 | 16.2 | 23.6 |
| None | 18.3 | 27.3 | 17.0 |
| 100.0 | 100.0 | 100.0 | |
| Employment Status | |||
| Unemployed | 26.7 | 34.4 | 26.7 |
| Employed | 32.8 | 22.7 | 19.7 |
| Full-time | 26.1 | 17.7 | 14.4 |
| Not in Labor Force | 40.5 | 42.8 | 53.7 |
| 100.0 | 100.0 | 100.0 | |
| Not in Labor Force | |||
| Homemaker | 9.1 | 12.6 | 8.8 |
| Student | 22.3 | 4.3 | 3.4 |
| Retired | 2.6 | 0.9 | 1.0 |
| Disabled | 18.9 | 15.0 | 23.1 |
| Inmate | 4.9 | 6.1 | 4.3 |
| Other | 42.3 | 61.0 | 59.3 |
| 100.0 | 100.0 | 100.0 | |
| Education | |||
| Less than HS Grad | 36.6 | 38.1 | 39.2 |
| HS Grad (or GED) | 42.9 | 41.6 | 42.7 |
| Source: TEDS, Office of Applied Studies, 1997. | |||
The Drug Abuse Services Research Survey (DSRS, Brandeis University, 1993) produced similar findings to those of the TEDS data and are arranged by site of care (Figure 12 below).
| Primary Payment Source (Phase I) | ||||
|---|---|---|---|---|
| No Payment | 15.5 | 28.6 | 17.6 | |
| Self Payment | 7.2 | 17.9 | 35.0 | |
| Private Health Insurance | 44.3 | 10.2 | 15.2 | |
| Medicaid | 12.0 | 8.2 | 14.6 | |
| Medicare | 4.0 | 0.1 | 1.1 | |
| Other Public | 17.0 | 35.0 | 16.5 | |
| 100.0 | 100.0 | 100.0 | ||
| Employed (Phase I) | 47.2 | 19.1 | 51.6 | |
| Educational Attainment (Phase II) | ||||
| Less than High School | 33.4 | 37.7 | 44.6 | |
| High School Graduate | 26.4 | 36.1 | 28.4 | |
| Beyond High School | 25.2 | 19.5 | 22.8 | |
| Unknown | 14.9 | 6.7 | 4.2 | |
| Age | ||||
| <18 years | 5.7 | 4.7 | 10 | |
| 18-24 years | 12.7 | 21.5 | 24 | |
| 25-34 years | 44.7 | 48.9 | 44.5 | |
| 35-44 years | 25.8 | 20.7 | 17.8 | |
| 45 + years | 11.3 | 4.2 | 3.8 | |
| 100.0 | 100.0 | 100.0 | ||
| Source: DSRS, Brandeis University, 1993a, 1993b. | ||||
Thus, the source and magnitude of funding for cocaine are key variables to consider in our market analysis. Payment for a new pharmacotherapy may come out of the $2.1 billion now being spent, from additional funds that could be made available, or from funds derived from a combination of existing and new sources.
The potential impact of the cost of a new medication for cocaine abuse may be perceived relative to the current cost of care. The wholesale price of certain commonly used psychotropic medications currently fall in the range of $2 to $4 per daily dose. If the wholesale price of a new cocaine medication is in that range, then its retail cost to payers would represent a large increase relative to current payments for cocaine treatment, particularly the average daily per-patient payment ($9.00) for the majority of cocaine patients in outpatient care (Figure 13 below). As for any health care intervention, the cost of a new cocaine pharmacotherapy should be considered in light of development costs as well as any health and economic benefits that may accrue from its use.
| Medication | |||
|---|---|---|---|
| Methadone | Opiate addiction | 50 mg | $0.50 |
| LAAM | Opiate addiction | 80 mg/2 days equal to 40 mg/day | $4.00/2 days equal to $2.00/day |
| Naltrexone | Opiate addiction, alcoholism | 50 mg | $4.50 |
| Diazepam | Antianxiety | 20 mg | $0.25 |
| Fluoxetine (Prozac) | Antidepressant | 40 mg | $4.50 |
| Chlorpromazine (Thorazine) | Antipsychotic | 600 mg | $3.00 |
| Haloperidol | Antipsychotic | 50 mg | $3.00 |
| Risperidone | Antipsychotic | 6 mg | $8.00 |
| Clozapine | Antipsychotic | 500 mg | $17.00 |
Source: 1996 Drug Topics Red Book. Medical Economics, Montvale, NJ, 1996. | |||
Psychosocial vs. Medical Treatment Model
Substance abuse treatment generally follows a psychosocial, rather than medical, model. The most recent surveys that have examined staffing patterns confirm that the substance abuse treatment system involves little or no physician time in the treatment of patients (Office of Applied Studies, 1993; Brandeis University, 1993). Even when on staff, physicians are often addressing primary health care needs or other mental disorders, rather than providing specialized substance abuse treatment services.
Methadone treatment for heroin addiction would appear to be the most medically oriented model of drug treatment. However, the role of physicians in methadone clinics is generally small and circumscribed to initial diagnostic assessments (i.e., of heroin addiction), management of methadone dosage, and some primary health care services. Most clinic services are oriented to the behavioral and psychosocial needs of the patients, and are delivered by counselors, social workers, and, less often, psychologists (Institute of Medicine, 1990).
The 1991 NDATUS study of specialty substance abuse providers surveyed 9,000 treatment centers (out of a total of about 11,500) with 811,000 patients enrolled on the survey date. The survey identified only about 2.2 full-time equivalent psychiatrists and other physicians, respectively, per 1,000 enrolled patients. Nationwide, there were about 88,000 full-time equivalent direct care staff, including about 1,800 psychiatrists and other physicians, respectively. These estimates were not adjusted for survey and item non-response (Office of Applied Studies, 1993).
The 1990 DSRS survey focused on 7,200 "drug" abuse centers (excluding "alcohol only" centers), serving 540,000 patients with primary or secondary drug problems. That survey found that there were about 1,000 full-time psychiatrists and other physicians, respectively, on staff at the 7,200 centers, very similar to the estimates of full-time physicians from NDATUS. The DSRS also found about 4,500 psychiatrists and other physicians working part-time or on contract; however, the report does not translate these numbers into full-time equivalents or into the proportion of facilities having any physicians on staff. These estimates were not adjusted for item non-response (Brandeis University, 1993).
Involvement of private practice physicians in treating cocaine addiction is virtually negligible. Of all national expenditures for specialized substance abuse treatment services, including alcoholism, less than 1 percent are for psychiatrist visits. Only about 1 percent of all visits to psychiatrists are by patients with a primary diagnosis of substance abuse, including alcoholism; another 2 percent are by patients with a secondary diagnosis of substance abuse (Harwood et al. 1994). Of all national expenditures for substance abuse services, excluding alcoholism, 2 to 3 percent are for visits to physicians of any type (Harwood 1984; Rice et al. 1990).
Comparison of Cocaine Market with Other Markets
The proportion of the entire population of heavy substance abusers that is considered to be a realistic target market for pharmacotherapy is small relative to the effective target market of pharmacotherapy for other diseases. Although there are other clinical conditions for which the market penetration of pharmacotherapy is proportionately low, the absolute magnitude of the overall market may be so large that even such small penetration of a substantially priced drug can be financially attractive to industry, as in the case of the smoking cessation market.
A combination of factors minimizes the attractiveness of the cocaine abuse market to industry, including: a relatively modest potential market (2.1 million heavy users), low proportion of users currently in treatment (250,000 enrollees on any given day), concerns about compliance in this population, and apparent market expectation of a low price point. This appears to be corroborated by the methadone market, where a relatively effective, low-priced medication in a well-established, long-standing treatment system achieves, at best, a 25 percent penetration of the small population of opiate addicts (i.e., about 125,000 patients enrolled in methadone treatment per day out of about 500,000 opiate addicts). The penetration of LAAM is less than five percent of the methadone market, i.e., less than one percent of the opiate addiction market, and the use of naltrexone for opiate addiction falls below that of LAAM. On the other hand, the market for medications to treat the 2.1 million epilepsy patients generates $400 to $500 million per year (IOM, 1995).
Drawing inferences about the potential market for a new cocaine abuse medication from the market experience of other medications for substance abuse must consider the market-limiting characteristics of the treatment systems for opiate addiction to which methadone and LAAM are subject. The market conditions for a new cocaine abuse medication would differ if it is provided via more traditional means of physician prescribing and distribution through pharmacies, rather than if it is a Schedule II or Schedule III controlled substance warranting the forms of controlled treatment delivery required for methadone and LAAM. Nevertheless, their status as medications for illegal substance abuse, similarities in the user populations, and other characteristics make methadone, LAAM, and naltrexone useful, though imperfect, market comparators for a new cocaine abuse medication. As a group, the markets for these medications more closely resemble the potential market for a cocaine abuse mediation than markets for other medications, and industry looks to these markets accordingly when assessing the market for potential cocaine abuse medications.
In comparison to other populations with clinically manifest cancer, heart disease, or other disorders, most substance abusers practice denial. Most abusers do not acknowledge a need for treatment and some even actively resist treatment. Many abusers that do acknowledge a need for treatment fail to seek or sustain treatment. Most patients that enter treatment do so under duress or coercion from legal authorities, employers, family members or friends (Institute of Medicine, 1990). Thus, social policy may be a primary driver of the proportion of cocaine addicts that can be directed to treatment in a year. However, such policies can be badly undermined when patients that do seek to enter treatment are greeted with waiting lists. Once placed on a waiting list, a patient is far less likely to be contacted and admitted when a treatment slot does open.
As noted in this report, it is estimated that there may be almost 900,000 treatment admissions or episodes per year by perhaps 800,000 cocaine addicts, although this number may be smaller due to high rates of relapse and treatment re-entry. Assuming that 800,000 users enter the system at least once during a year, this would constitute nearly 40 percent of the total number of heavy cocaine users, and might appear to be a more substantial market. However, the prospects of retaining this number of patients in ongoing treatment do not appear favorable, given the limitations of a treatment system and characteristics of a population that yield only 250,000 daily enrollees.
The uncertainty about the number of cocaine users, especially the secondary users, places a wide confidence interval around the potential size of the market for cocaine abuse treatment. For the purposes of gauging the potential market for a new pharmacotherapy for cocaine addiction, it may be optimistic to use an estimate of 250,000 current daily enrollees in treatment. Projections of market penetration must consider that some primary users and many secondary abusers may be treated for other addictions and with behavioral therapy to the exclusion of pharmacotherapy, particularly in the context of the current treatment system. Given that many cocaine addicts abuse multiple substances and have diverse health and behavioral disorders, it may be that one or a few medications for cocaine abuse will be insufficient for treating this population. To the extent that multiple medications are needed, the market potential for any one medication would be reduced.
From the standpoint of the pharmaceutical industry, the anti-depressant drug market may serve as a useful contrast in the CNS market to the cocaine market. Depression is a condition that afflicts more than 8 million Americans (Irvin 1997), and depressive disorders have a lifetime prevalence of up to 15 percent for men and 24 percent for women (Hirschfeld 1997).
The consequences of depression include patient mortality as well as direct and indirect costs to society. Those that suffer from depression are at a greater risk for suicide and other co-morbidities including substance abuse, heart disease and other medical conditions. In 1990 alone, the cost of depression was estimated to be between $26 and $43.7 billion (Henry 1997). On average, employers incur an annual cost of $4,200 per depressed employee, of which 28 percent represents treatment costs (Hirschfeld 1997).
In terms of utilization and sales of anti-depressant drugs, 98 million prescriptions were written for these drugs and $4.3 billion in total sales were recorded in 1996 alone (Scott-Levin 1996, IMS 1996). The selective serotonin re-uptake inhibitors (SSRIs) were the most prescribed anti-depressant (54 percent) and captured 89 percent of the market dollars, while the tri/tetracyclics accounted for 5.6 percent of the market dollars despite representing 32 percent of all anti-depressant prescriptions.
The vast majority of these anti-depressants are dispensed through the retail sector (IMS 1996). Retail sales (i.e., through chain stores, independent pharmacies, food stores) accounted for 86 percent of the market in 1996, while long term care, federal facilities, HMOs, non-federal hospitals, and clinics made up the remaining 14 percent of the market.
Price Sensitivity of a Cocaine Medication
Since no approved pharmacotherapy for cocaine abuse has been tested on the market, it is not possible to know how sensitive the market would be to such a medication. However, indirect available evidence from other substance abuse medications and the current nature of cocaine abuse treatment and its financing would appear to indicate that the market would be very sensitive to the price of a cocaine medication.
In a market where the average daily treatment cost is a modest $9.00 per patient for standard outpatient care (accounting for the great majority of patients) and $23.00 per patient across all modalities of care, a cocaine pharmacotherapy priced at a daily dose of a few dollars would represent a significant proportionate cost increase. This may be particularly so in the judgment of substance abuse treatment providers that are vested in psychosocial approaches to the exclusion of pharmacotherapy. It is important to note that the price sensitivity of the current treatment system may vary considerably from that of more typical pharmacotherapy markets that involve physician prescribing and distribution through pharmacies.
The price of methadone may exert some pull on the price point for a cocaine medication. The price for that relatively effective medication, used to treat another stigmatized substance abuse population and paid for primarily by government sources, is a mere 50 cents per daily dose. Although the price of medications for smoking cessation is considerably higher, such medications are not paid for out of public sources, but rather by a self-pay population.
As noted in the case study of LAAM (see Case Studies section), in certain states, Medicaid has reimbursed clinics for the cost of methadone (about $0.50 wholesale per daily dose) but would not cover the higher, though still modest, cost of LAAM (about $2.00 wholesale per daily dose). In such instances, clinics either had to negotiate with the state to receive greater funding or had to absorb the additional cost associated with LAAM. Market resistance to naltrexone, which at about $4.00 wholesale per daily dose is twice as expensive as LAAM yet moderately priced compared to other drugs, was complicated by the additional cost of needed adjunctive psychosocial services. As such, state programs cannot afford to treat populations of heroin addicts with naltrexone ($3,500 per patient annually), which carries double the cost of treatment with methadone ($1,200 to $1,700).
Most existing treatment for cocaine abuse is paid for out of fixed annual government appropriations. Treatment centers that must function with capped budgets and that are judged on the number of patients treated would be expected to be price sensitive. Traditional measures of treatment productivity have been the number of patients treated or the cost per patient treated, rather than measures of improved health outcomes. In the current environment, apparent productivity can be increased by reducing the cost per person served, as occurred in treatment funding from the 1970s to the 1980s (Institute of Medicine, 1990). The introduction of any additional and/or more expensive services reduces the number of patients that can be served with a given budget. In the current treatment and financing context, it is likely that treatment systems and providers will be very sensitive to the price of any new cocaine pharmacotherapy.
Basic Relationships of Price, Market Size, and Revenues
As described above, on any given day there are 250,000 cocaine patients enrolled in treatment, out of an estimated 2 million cocaine addicts in the U.S. If a new medication were used by every current patient in treatment, the medication would have to sell at just over $2.00 per daily dose wholesale ($2.20 to 4.00 per day retail) in order to generate $200 million in annual revenue, as shown in Figure 14 below. If the peak market penetration is only 50 percent of current patients, the wholesale price will have to be over $4.00 per daily dose. On the other hand, if the number of cocaine patients in treatment can be doubled, the necessary wholesale price would only be about $1.10 per daily dose. In light of the importance of achieving significant market penetration, it is important to note that for the drugs LAAM and naltrexone, market penetration in the first several years has not reached 5 percent (see Case Study section).
Decision makers in industry must consider:
- how many patients would use a cocaine medication (as modulated/governed by their providers)
- how sensitive demand will be to price
- how much payment will be forthcoming for the added cost of medications.
If PAR is substantially below $200 million, or some other acceptable industry threshold, there may still be commercial interest if there is reason to expect a sufficient NPV. This will depend primarily on costs of development and production relative to the expected lifecycle of revenue. In order to elicit significant pharmaceutical company development interest at revenue levels below $200 million, the expected costs of development probably would have to be lower than for a "typical" medication. According to 1994 estimates by the former congressional Office of Technology Assessment (OTA), it cost an average of about $150 million in cash outlays, and a capitalized cost in excess of $350 million over 12 to 15 years to develop a successful new medication, including the costs associated with development of drugs that never reached the market. The cash outlays for any given drug that did reach the market, not including the burden of cash outlays for drugs that did not ultimately reach the market, were generally less than $50 million per successful drug in the OTA report.
Figure 14: Wholesale Price of Daily Medication Needed to Yield $200 Million in Annual Revenue, by Number of Daily Clients
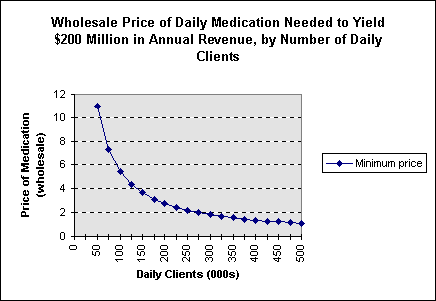
Translating Market Barriers and Policy Options into Financial Parameters
As described in this report, pharmaceutical companies' decisions to pursue new products is based largely on financial considerations inherent in risk-reward tradeoffs. Despite the great diversity of market barriers, most can be interpreted as having a direct effect on one or more financial parameters that are factored into these decisions. Similarly, most policy options that exist or that could be implemented can be interpreted as having a direct effect on these financial parameters.
The figures below portray likely relationships between the particular market barriers (Figure 15) and policy options (Figure 16) identified in the 1995 IOM report and six basic parameters relevant to decisions to pursue a new therapy: R&D costs, (time to) product launch, marketing and distribution costs, market size/penetration, price, and (duration of) market life. These relationships were used in part to develop the scenarios shown later in this report. In particular, these relationships were used to translate market barriers, and various means that might lower these barriers, into the financial parameters used to generate the quantitative indicators of PAR and NPV. (Note: the use in Figures 15 and 16 of the market barriers and policy options cited in the IOM report does not imply that this study confirms all of those market barriers and policy options.)
Figure 15: Effects of Market Barriers on Types of Financial Parameters: New Medications for Substance Abuse (barriers drawn from IOM report)
| R&D Costs | Product Launch | Marketing / Distribution Costs | Marketing Size / Penetration | Price | Market Life | |
|---|---|---|---|---|---|---|
| Discovery | ||||||
| Limited number of researchers focusing on drug abuse | X | X | ||||
| Lack of well-characterized animal models of cocaine addiction | X | X | ||||
| Limited basic science knowledge of addiction, craving, and relapse | X | X | ||||
| File IND | ||||||
| Clinical Studies | ||||||
| DEA regulations | X | X | ||||
| Complications of concomitant illness and polydrug abuse | X | X | ||||
| Patient population perceived as difficult to study | X | X | ||||
| Efficacy outcomes difficult to define or measure | X | X | ||||
| Few clinical investigators | X | X | ||||
| File NDA | ||||||
| Length of FDA approval process | X | X | X | |||
| Other Approval | ||||||
| State rescheduling | X | X | ||||
| Varied state / local approval processes | X | X | ||||
| DEA review time | X | X | ||||
| Marketing | ||||||
| Varied state and local regulations | X | |||||
| Lack of traditional marketing to physicians | X | |||||
| Pricing clause in DHHS CRADAs* | X | |||||
| Small foreign market | X | |||||
| Treatment System | ||||||
| Limited number of narcotic treatment programs | X | |||||
| Stigma of drug-abuse | X | |||||
| Bias by some treatment providers against pharmacologic treatments | X | |||||
| Varied state / local treatment regulations and financing mechanisms | X | X | ||||
| Uncertain treatment financing | X | X | ||||
Market barriers derived from IOM (1995). *No longer applicable. | ||||||
| R&D Costs | Product Launch | Marketing/Distribution Costs | Market Size/ Penetration | Price | Market Life | |
|---|---|---|---|---|---|---|
| Improve funding for government medications development | x | |||||
| Designate and support national drug abuse research centers | x | x | ||||
| Establish / Enhance Leadership in Improving Relationship w/ Industry | ||||||
| Expand Treatment Capabilities of States | ||||||
| Provide more funding to increase treatment where there are waiting lists | x | |||||
| Shift funds from supply control programs to treatment programs | x | x | ||||
| Treatment Financing | ||||||
| Require all Substance Abuse Block Grant recipients to offer approved anti-addiction medications | x | x | ||||
| Assure appropriate financing of new medications by state alcohol and drug agencies and their counterpart Medicaid agencies | x | x | ||||
| Training and Education | ||||||
| Increase government training budgets | x | x | ||||
| Regulatory Policies (FDA) | ||||||
| Make treatment-IND, parallel track, and accelerated approval available for anti-addiction medications | x | x | ||||
| Regulatory Policies (DEA) | ||||||
| Count DEA review time as part of regulatory process for purposes of patent term extension for controlled substances | x | |||||
| Remove / reduce bureaucratic burden on clinical investigations involving controlled substances | x | x | x | |||
| States | ||||||
| Begin scheduling process as soon as possible after submission of NDA | x | |||||
| Work more closely with states to prepare path for new anti-addiction medications | x | x | ||||
| Compile information about state regulatory processes and educate state regulators and pharmaceutical company representatives | x | x | ||||
| Develop a series of specific actions encouraging states to reform their laws and regulations to facilitate availability of new anti-addiction medications that are controlled substances | x | x | x | |||
| Draft (national) legislation requiring states to implement needed changes | x | x | x | |||
| Market Obstacles and Creating Incentives | ||||||
| Size of Market | ||||||
| Grant orphan drug (or similar) status to FDA-approved anti-addiction medications whose potential market can reasonably be judged to meet the 200,000 patient criterion | x | x | ||||
| Drug Pricing and Intellectual Property Rights | ||||||
| Remove or modify reasonable pricing clause for CRADAs | x | |||||
| Streamline CRADA process | x | |||||
| Societal Stigma | ||||||
| "Need for national leadership in support of pharmacotherapy and continued emphasis on prevention and treatment. The sense of social stigma is most likely to diminish as a result of public education and broader acceptance of addiction as a treatable disease." | x | x | ||||
| Need for Federal Leadership | ||||||
| Executive order assigning a high priority to the development of medications for drug-abuse treatment (or some other explicit action) | x | x | ||||
| Options for Further Consideration | ||||||
| Offer developers of the first few FDA-approved mediations, for three years after approval, a federal subsidy of a maximum of $50 million for purchase of the drug (e.g., via reimbursement of copayment portion of medications for patients with health insurance and the full cost of medications of patients without health insurance) | x | |||||
| Standing federal purchase orders for prearranged quantities at an adequate price for one or more new cocaine treatment medications to begin at FDA approval | x | x | ||||
| * Policy options derived from IOM (1995). | ||||||
Modeling Financial Return
The level of interest on the part of industry in developing a new drug abuse medication depends primarily on financial criteria. Factors that may not appear to have direct financial import, such as social stigma associated with a product, inclination of caregivers to consider pharmacological as opposed to behavioral interventions, or corporate commitment to further the greater societal good, do have financial implications that are considered by companies. The financial impacts of such factors can be estimated and incorporated as such into decisions about pursuing projects.
Companies often have summary financial targets or hurdles that drive investment decisions. Two that are often used in industry and which are used in this report are net present value (NPV) and peak annual revenue (PAR). These and other related terms are defined in Figure 3 (earlier in the Methods section).
In principle, companies pursue projects that have positive NPVs. In addition, larger companies are less likely to be interested in a new drug for which projected PAR is less than $200 to 300 million. Of course, alternative projects that offer higher NPVs and/or PARs tend to be more attractive to companies.
Gauging whether a medication might generate a given level of annual revenue to a company is relatively straightforward, as it depends primarily on the number of patients taking a given amount of medication per day and the wholesale price (i.e., paid to the company) per day of the medication. Figure 17 (below) presents this fundamental relationship.
Figure 17: Peak Annual Revenue by Peak Daily Patients, at 3 Wholesale Prices
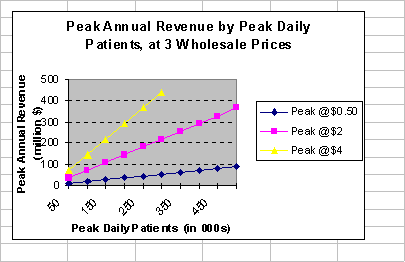
Source: Analysis by The Lewin Group
As shown in Figure 17, a new cocaine medication could achieve $200 million in revenue with 150,000 daily patients (i.e., who are on a prescription on any given day) and a wholesale medication price of $4.00 per day, or with 250,000 daily patients and a wholesale price of about $2.00 per day. In order to achieve $200 million at a wholesale price of $0.50 per day, there would have to be more than 1 million daily patients. The graph can be interpolated or extrapolated to estimate the number of patients that would be required at other prices in order to achieve an annual revenue of $200 million.
Market Penetration, Price, and Annual Revenue
In order to demonstrate basic relationships between market penetration, price, and annual revenue, the following analysis poses three levels of market penetration and three price levels for a new cocaine medication. The market penetration levels are "doubling," "saturation," and "half entry," as follows.
- Doubling the current number of cocaine abusers in treatment to 500,000 patients per day might be achieved if a national program for long-term maintenance treatment was developed. Such a level of market penetration would be equivalent to that of methadone with opiate addicts, i.e., penetration of approximately 25 percent of heavy users.
- Saturation of the current 250,000 patients per day (i.e., the current number of patients plus recently discharged patients) would be a high rate of market penetration. This would involve acceptance of patients for medium-term treatment (3 to 6 months), though not for indeterminate duration maintenance.
- Half entry of 50 percent of the current daily census of cocaine users in treatment would yield 125,000 patients. This would involve medication accepted for use while patients were under care of specialty or health providers (for an average of about 3 months), but not after termination of formal treatment.
These levels of market penetration and service delivery are patterned on currently existing service configurations in the national treatment system. As such, these levels are based on the estimated number of patients currently enrolled in treatment (i.e., 250,000), rather than on the estimated number of cocaine users that have been treated once or more in a given year (i.e., 900,000).
The market penetration levels are linked to payment levels in order to derive variables of financial return to pharmaceutical companies. For purposes of illustration, three pricing levels are as follows.
- $2.50 wholesale per daily dose
- $1.25 wholesale per daily dose
- $0.50 wholesale per daily dose
The wholesale price of $2.50 per daily dose is lower than that of naltrexone, a medication for both opiate addiction and alcoholism, which is about $4.55 per daily dose. The wholesale price of $0.50 is used as a lower price limit; it is roughly what methadone programs pay for their medication. (It is unlikely that a new pharmacotherapy for cocaine abuse would be priced so low.) The price of $1.25 is halfway between the other two, and is near the low end of the price range for LAAM. As a point of reference, providers pay about $1.30 - $2.00 per daily dose of LAAM (i.e., 180 - 280 mg/week at $0.05/mg.).
Estimated revenue to a company is based on the wholesale prices, i.e., the price per dose paid by providers (e.g., pharmacies or drug treatment clinics, depending on how a drug is dispensed) to the pharmaceutical company. However, the retail prices (i.e., that cover the cost of medication plus related costs of storage, handling, and dispensing) that are charged to patients and/or that are passed through to payers (insurance companies, Medicaid programs, other payers) are relevant as well, as any payer reluctance to pay these retail amounts may affect use of a drug. Retail markups vary widely, but are typically on the order of 10 - 100 percent.
In the current context of treatment, a new cocaine medication would represent additional payments at retail prices paid by third-party payers (government or private sector), patients, or other sources. The estimates for retail prices for medication should be considered relative to their anticipated net impact on current spending levels for treatment noted above, i.e., an average of $23.00 per day across all 250,000 enrolled cocaine users and an average of $9.00 for the majority of cocaine users enrolled in outpatient programs. At a wholesale price of $4.00 per day, a retail markup for a typical Medicaid program of 15 percent would yield a retail price of $4.60. Assuming that the cost of the medication would add to, and not substitute for, the current cost of care of $9.00 per day, the cost of the new medication would represent to the payer a 51 percent increase in the daily cost of treatment of cocaine abusers. In comparison, at $0.50 per day, the cost of methadone is only a small portion of the $5 to $20 daily cost of treating opiate addicts.
Peak Annual Revenue
Figure 18 (below) shows expected peak annual revenue to a pharmaceutical company under the nine combinations of market penetration and wholesale price. Under the most optimistic combination of these, the PAR to the pharmaceutical company would be $455 million, well above most companies' acceptable PAR thresholds. However, achieving this PAR would require the following.
- There would be 500,000 cocaine users (double the current 250,000) enrolled in treatment daily, representing about 20 to 25 percent of the estimated total number of heavy cocaine users.
- Payers would be willing to pay roughly $2.75 to $5.00 retail for medication per day per enrolled user.
- Approximately $500 to 600 million of funding for treatment with the new drug (i.e., the retail payments for $455 million wholesale of the drug). This would have to be realized in the form of new funding, reallocation of funds from the $2.1 billion currently spent for treating the 250,000 patients currently enrolled in treatment, or a combination of these.
Achieving these assumptions would likely require creation and funding of a new national substance abuse treatment program for cocaine users. Precedent for such new funding exists in the creation of the national methadone maintenance treatment system in the early 1970s, when nearly 100,000 methadone treatment slots were funded de novo. This was equivalent to 20 to 25 percent of the estimated number of heroin addicts at that time (also the current penetration of methadone treatment). This major funding initiative was supported primarily by the federal government and entailed, as planned, a phase-down of federal dollars over the subsequent five years.
| Penetration | ||||
| Doubling | $455.0 | $227.5 | $91.0 | |
| Saturation | $227.5 | $113.8 | $45.5 | |
| Half entry | $113.8 | $56.9 | $22.8 | |
A PAR of $227.5 million could be achieved through: 1) the doubling of current market penetration of cocaine treatment (likely requiring a national cocaine maintenance system) with the middle-tier price or 2) current treatment levels with the higher price. Another way to realize this estimate would be if two-thirds of current daily patients and about one-third of the recent departures from treatment took the medication. Under this scenario, patients would take the medication for an average of three months during treatment, and half might continue for another three months. (These average times for taking medication are illustrative only. Such determinations would necessarily reflect such factors as whether a drug is intended for short-term use, e.g., for detoxification, or indefinite maintenance, as well as patient drop-out rates, which can be considerable.) As noted above, achieving this estimate would require a major infusion or diversion of resources, equal to about 15 percent of current funding for treatment of cocaine abusers.
A PAR of $100 million per year could be achieved under more moderate, though still ambitious, scenarios. Market penetration of 50 percent would require a wholesale price of $2.50 per day to generate this level of revenue. If a new medication reached the current number of daily enrollees, the price can be about half as high, i.e., $1.25 wholesale. Doubling the current number of treated patients would yield $91 million per year at a retail price of only $0.50 per day.
The time to achieve PAR reflects the level of investment in a new drug, and affects the number of years during which peak annual revenues can be sustained, i.e., before a product loses its market exclusivity. This is addressed in the sample scenarios later in this report.
Internal Rate of Return and Reducing the Time to Market
Another important investment criterion used by some companies is the internal rate of return (IRR). The IRR is the rate of interest at which the present value of all net cash flows into and out of a project over a specified time interval equals zero. A higher IRR makes an investment opportunity more favorable.
According to its report on the pharmaceutical industry, the former congressional Office of Technology Assessment (OTA) estimated that the pharmaceutical industry achieves IRRs in the range of about 14 percent (OTA 1993). To maintain such a rate, companies may seek individual investment opportunities with higher rates, e.g., above 20 percent, to account for the risk associated with drug development efforts, the majority of which do not ultimately yield marketable medications.
Consideration of acceptable IRR for prospective cocaine medications readily reveals two key findings. First, if the cost of developing a successful cocaine medication is comparable to OTA estimates of the recent cost of developing other medications, pharmaceutical companies will be hesitant to take on such efforts early in the R&D process. OTA estimated that the fully capitalized cost of a medication at launch was as much as $359 million in 1990 dollars, equivalent to cash outlays of $135 million over 13 years. When these factors are applied to the market scenarios described above, not even the optimistic doubling scenario at the high wholesale price of $2.50 per patient per day meets the 20 percent IRR threshold, let alone the more conservative saturation and half-entry scenarios.
Second, pharmaceutical companies may be more interested in a late-entry scenario in which a company takes on further development after initial R&D - and its inherent risk - have been conducted (or sponsored) by another entity. Late entry might occur if, as has happened with other medications, the government were to complete the early years of product development and then turn over the rights of the product to a company.
As indicated in Figure 19 (below), a late-entry scenario that requires only 8 years to complete development of a cocaine medication, at an estimated total cash outlay of $70 million, may be more favorable. This scenario, entailing a shorter development time at lower cost, may be attained when the government, another company, or some combination of these has invested in early R&D. Thus, a large pharmaceutical company may be more willing to acquire or otherwise collaborate with a smaller firm with a substance abuse compound that has already passed milestones in the R&D pipeline, requiring less investment and fewer years to market. This scenario may also occur when a major advance in the science base has occurred that accelerates the drug discovery and development process. New research indicating that medications in development for other central nervous system disorders (mental illness, analgesia, anesthesia, etc.) have applications for substance abuse as well could shorten the development time table and lower costs.
| 8 yrs / $70 million | ||||
| Doubling | $455 mil. | 17.6 | 27.5 | |
| Saturation | $227.5 mil. | 13.0 | 21.4 | |
| Half Entry | $113.8 mil. | 8.4 | 15.4 | |
| Assumes, e.g., $2.50 wholesale cost per day for medication, 4 years from launch to peak sales, patent acquired 3 years after start of development. | ||||
Note that even the least ambitious penetration market scenario achieves the 20 percent IRR threshold value if the cost of the development phase is sufficiently reduced, Figure 20 (below).
| $50 million | $30 million | ||||
| Penetration | |||||
| Doubling | $455 mil. | 27.5 | 30.7 | 35.2 | |
| Saturation | $227.5 mil. | 21.4 | 24.5 | 28.9 | |
| Half Entry | $113.8 mil. | 15.4 | 18.4 | 22.7 | |
| Assumes, e.g., $2.50 retail cost per day for medication per patient, 8 years of development until launch, 4 years from launch to peak sales, and 14 years of patent life, post-approval. | |||||
Affordable Levels of Development Cost
The final approach taken to assessing the financial attractiveness to commercial firms is to estimate the affordable level of R&D cost, given the expected peak annual revenue to the company. The base conditions for the model have been used to develop the estimates in Figure 21 (below). In particular, this set of calculations assumes that a company has a potential medication that has completed preclinical development, and only has an expected 8 years until product approval and launch.
The graph represents, for a given PAR, the maximum level of expected development cost that would allow a firm to anticipate a 20 percent IRR. For example, if the acceptable threshold for peak annual revenue of a cocaine medication is as low as $50 million, the maximum anticipated cash outlay would have an expected value of $18 million over 8 years. The expected value takes into account the risk of failure of the development effort; thus, the actual outlays could be as low as $5 - 6 million for a single potential compound.
Figure 21
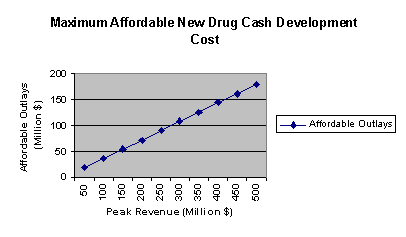
In this model, only at a PAR near $400 million would a pharmaceutical firm expect a "typical" development effort with cash outlays (adjusted for risk of failure) to have good prospects of yielding a strong payoff. Below $400 million per year, the company would have to either anticipate lower than average development costs, or a higher than average probability of a successful development effort.
Earlier in this report it was suggested that a major pharmaceutical company would want to anticipate a potential peak market of $200 million in order to seriously consider a development project. At this level of expected revenue the company would still have to have very positive prospects for development of a medication in order to proceed. Either there would have to be reason to believe that costs could be kept lower than for an "average" medication development effort, or they would need to anticipate a higher probability of success. These calculations are predicated on the assumption that the annual revenue target will be achieved through successful entry into the market at an adequate price.
Scenarios of Company Decision Making
This report presents several scenarios of pharmaceutical company decision making regarding undertaking projects to develop pharmacotherapies for substance abuse (e.g., cocaine) under various sets of market conditions. Using a quantitative model developed by The Lewin Group, the market conditions are translated into financial and other parameters to generate estimated PAR and NPV for each scenario and for certain variations of these scenarios. Modeling these scenarios helps to illustrate some of the key barriers and other limitations to development of medications for cocaine abuse, as well as the extent to which certain types of financial and policy options might reduce such barriers. Policy options to lower barriers include some that already exist and some that have been posed by the IOM (1995).
The market conditions of each scenario are translated into parameters that are fed into the model to generate estimates of PAR and NPV. Four of the base scenarios have multiple variations, or sub-scenarios, to explore the sensitivity of the output measures to changes in selected parameters. The values assigned to most of the parameters for each base scenario are shown at the end of each scenario.
The five main scenarios are as follows.
Scenario 1: Big Pharm Cold Start. A large pharmaceutical company considers taking on a full product development cycle for a new medication. Sub-scenarios explore how changes in competition, pricing, and market penetration would affect PAR and NPV.
Scenario 2: Biotech Gets Help. A small biotechnology firm takes on the full product development cycle with a promising compound, given the expectation of a set of government incentives to pursue the product. Sub-scenarios explore the sensitivity of PAR and NPV to the various government incentives.
Scenario 3: Guaranteed Handoff. A pharmaceutical company considers accepting a government offer for the rights to a product that the government has taken into phase III clinical trials in the context of additional government incentives.
Scenario 4: Vaccine. A pharmaceutical company considers accepting a government offer for the rights to a product entailing a vaccine and annual booster that the government has taken through preclinical work, in the context of additional incentives provided by the government.
Scenario 5: Second Indication. A pharmaceutical company has a highly successful product that is already approved for an existing CNS indication. The product shows promise for treatment of cocaine addiction as a second indication. The company considers whether to pursue this second indication.
These scenarios are for discussion purposes only. They are illustrative, and not exhaustive, of the possible combinations of market conditions used here. The scenarios are entirely hypothetical works drafted by The Lewin Group. The scenarios do not represent any suggestion or intent by the government to adopt any policies described here. Certain policy options posed here are already in existence, e.g., orphan drug or similar status; others have been discussed in the 1995 IOM report cited in this document.
Scenario 1: Big Pharm Cold Start
In this scenario, a large pharmaceutical company takes on the full product development cycle, i.e., a "cold start" with preclinical discovery and research. The new Product A will require about $150 million in uncapitalized expenditures to develop and will be ready for launch following FDA approval in 13 years. The company anticipates securing a patent 4 years into the 13-year development process, about the time it expects to start phase I clinical trials. The company has a strong CNS product line, and takes a confident view about its chances for success, and therefore uses its typical cost of capital figure of 12 percent.
It will take 5 years post-launch for Product A to reach its peak market of 125,000 daily enrollees, i.e., 50 percent of the current 250,000 daily enrollees in treatment for cocaine abuse. The drug will be priced at $2.50 per daily dose, comparable to the current price of LAAM. The average enrollee will take 13 weeks worth of prescriptions.
A competing drug (not a generic version of Product A) will be in development that will appear on the market 3 years after launch of Product A, and will completely replace Product A on the market after 10 years.
The company is highly reluctant to invest in projects that yield a PAR less than $250-300 million. Although in principle it would pursue project that has a positive NPV, the company is reluctant to pursue projects with NPVs of less than $100 million.
Base scenario. Under the base scenario described above, the company determines that the PAR of Product A would be $102 million, and that the NPV of investing in Product A would be a loss of $52 million. Given the unfavorable indicators of this basic scenario, the company explores the potential effects of certain variations, as follows.
Base + orphan. Given the relatively small size of the current market of people in cocaine abuse treatment, the company considers that Product A could be granted orphan drug (or similar) status. Given that the drug's patent would not expire until more than 7 years after approval, orphan status would not confer additional market protection (including against the competing drug noted above), but it would provide tax credits for qualifying clinical R&D expenditures, assumed here to be about 20 percent of such expenditures. Under this revised scenario, the company determines that the PAR of Product A would remain at $102 million, and the NPV would be a loss of $36 million.
Base + orphan + late competition. The company determines that, if no competing drug (whether a generic version of Product A or another competing drug for the same indication) were to appear on the market for 10 years post-launch, and such a competing drug took another 10 years to overtake Product A on the market, then the PAR would rise slightly to $114 million and the NPV would still be a loss of $15 million.
Base + orphan + late competition + achieve $300 million PAR. The company seeks to determine what price would be required to achieve a PAR commensurate with the company's target for a new drug. Assuming orphan-like status, late entry of any competitor, and 50 percent market penetration as above, the company determines that the wholesale price of the drug would have to be $6.60 per daily dose in order to achieve a PAR of $300 million, with an NPV of $60 million. The company notes that this wholesale price per daily dose is two-to-three times that of LAAM and about 50 percent higher than that of naltrexone. Although price may be only one of multiple contributing factors, company analysts note that both LAAM and naltrexone have fallen far short of penetrating their target markets at 50 percent, as is assumed in this scenario.
Base + orphan + premium price. The company seeks to determine what effect premium pricing would have on its financial outputs. For an upper bound, executives note that payments for triple therapy for HIV/AIDS patients can be $10,000 to $15,000 per patient per year. Thus, assuming orphan status and making the outlier assumptions of payments of $10,000 per patient per year (equivalent to $27.40 per patient per day and perfect compliance all year) and 50 percent market penetration of 125,000 daily patients, the company determines that its PAR would be $1,122 million, with an NPV of $209 million. At the same premium price of $27.40 and other assumptions retained, the company determines that it could achieve a PAR of $300 million with a market penetration of about 33,000 patients, or about 13 percent of the current 250,000 daily enrollees.
Base + low penetration. Some company analysts insist on taking what they consider to be a more realistic view of the potential market. They cite the low market penetration to date of LAAM and naltrexone, the lack of assurance of securing orphan status, and the lack of assurance that no viable competition would enter the market for a full decade after launch of Product A. Assuming a wholesale price of $2.50, peak penetration of just 10 percent of the 250,000 current daily enrollees, no orphan status, and appearance of a competing drug 3 years after launch, they determine that PAR would be $20 million and the NPV would be a loss of $71 million.
The company determines not to go forward with a cold start for a new medication for cocaine abuse.
Key Parameters (base scenario):
Time to patent expiration: 21 yrs.
Time to product launch: 13 yrs.
Premarket R&D expenditures: $150m
Cost of capital: 12%
Orphan drug/similar status: no
Post-launch to peak prescriptions: 5 yrs.
Peak daily patients: 125,000
Average weeks prescription per year: 13 weeks
Average daily dose wholesale price: $2.50
Time post-launch to competing drug: 3 yrs.
Time for competing drug to replace: 10 yrs.
Scenario 2: Biotech Gets Help
In this scenario, a small biotechnology firm, Company B, takes on the full product development cycle with a new class of compounds based on a recent scientific breakthrough that is highly specific to cocaine receptor neuropharmacology. The company anticipates that a new Product B from this class of compounds will have excellent compliance in the user population.
Company B is confident that Product B would require only about $50 million in uncapitalized expenditures to develop over a typical 13-year period to FDA approval. The company is backed considerably by venture capital, and must use a discount rate of 15 percent, higher than the figure of 12 percent typically used by many larger pharmaceutical firms. Analyzing the competition in the field, Company B estimates that a competing drug that is not a generic version of Product B will enter the market some 7 years after the launch of Product B and will take another 10 years to completely usurp Product B in the market.
Aside from the breakthrough nature of the new product, Company B's optimism is grounded largely on three pending government policies that are "highly likely" to be implemented, as follows.
The first policy is a pending regulatory reform that would effectively reduce the time to market approval by 1 year. This means that Product B would be on the market a year earlier, i.e., in 12 years rather than 13. As such, Product B would also have another year on the market before entry of the competing non-generic product noted above.
The second policy concerns market protection, i.e., providing orphan drug-like status for cocaine medications for indications of populations no larger than 300,000 (as compared to the current figure of 200,000 used for orphan drug status). Company B realizes that such status would provide protection from a generic substitute for 7 years post-launch; however, the company anticipates that this will not affect Product B, whose patent will not have expired during that period. Orphan-like status would provide tax credits for qualifying clinical trial expenditures, assumed to be about 20 percent of R&D expenditures.
The third policy is a federal commitment to expand treatment and financing capabilities at the state level. This commitment would involve the following elements: (a) provide more funding to increase treatment capacity, (b) require all substance abuse block grant recipients to offer approved anti-addiction medications, and (c) assure appropriate financing of new medications by state alcohol and drug agencies and their counterpart Medicaid agencies.
With the new provisions at the state level, Company B is confident that the total number of people seeking treatment will increase substantially, and the proportion of those that will be daily enrollees in treatment per year will increase as well. The company assumes it will take 5 years post-launch for Product B to reach its peak market of 250,000 daily enrollees, equal to the current number of daily enrollees in treatment for cocaine addiction.
The company assumes that the drug will be priced at $2.50 per daily dose, comparable to the current price of LAAM. The average enrollee will take an average of 26 weeks worth of prescriptions per year.
Company B and its backers want a reasonable chance of having a product that will attain a PAR of $200 million and a positive NPV.
Base scenario. The company determines that the PAR of Product B would be $228 million, and that the NPV (at a 15 percent discount rate) of investing in Product B would be $36 million.
Cautious investors note that Company B's determination depends on the three government interventions, as well as an assumption of 100 percent penetration of the current market of cocaine addicts in treatment. These investors want to know how the absence of each intervention would affect PAR and NPV.
Base with no regulatory reform. If the regulatory reform is not realized, Product B will require 13 years to market approval, and the competing non-generic drug will enter the market a year earlier relative to the launch of Product B. The PAR would remain at $228, but the NPV would decrease to $25 million.
Base with no orphan-like status. If market protection is not given based on the higher population criterion, but the other assumptions remain, the resulting loss of tax breaks would decrease the NPV moderately, from $33 million to $28 million. The PAR would remain at $228 million.
Base with no state provisions. In the absence of the commitment to expand treatment and financing capabilities at the state level, but the other assumptions remaining, the market penetration would likely fall far short of the projected 250,000 daily enrollees. If it fell to a still substantial level of 125,000 daily enrollees (50 percent of the current number of daily enrollees in treatment for cocaine abuse), the PAR would be $114 million and the NPV would drop to $7 million.
Base with low penetration. Assuming the provisions of the base scenario but reducing the market penetration to 25,000 (10 percent of the current daily enrollees in treatment for cocaine addiction), the PAR drops to $23 million and the NPV is a loss of $18 million.
Although the company remains optimistic about pursuing Product B, investors seek additional assurances regarding the government initiatives, particularly the state initiatives, and seek more information about the prospects for significant market penetration.
Time to patent expiration: 20 yrs.
Time to product launch: 12 yrs.
Premarket R&D expenditures: $50m
Cost of capital: 15%
Orphan drug/similar status: yes
Post-launch to peak prescriptions: 5 yrs.
Peak daily patients: 250,000
Average weeks prescription per year: 26 weeks
Average daily dose wholesale price: $2.50
Time post-launch to competing drug: 8 yrs.
Time for competing drug to replace: 10 yrs.
Scenario 3: Guaranteed Handoff
In Scenario 3, a government research agency has taken a highly promising compound, Product G, into Phase III clinical trials. The government approaches a large pharmaceutical firm, Company G, that has a strong CNS product line. The government offers to turn over the rights to Product G in exchange for assistance in completing Phase III trials, securing FDA approval, and conducting an active marketing campaign through at least 5 years post-launch. The patent on the drug will expire in 3 years, at about the time the product is expected to be launched. Contingent on FDA approval and the marketing campaign, the government will: (a) award orphan drug-like status, providing 7 years of post-launch protection against generic competition, (b) add an additional 5 years of protection against generic competition at the end of the orphan period, (c) guarantee purchases at a wholesale price of $2.50 per daily dose for Product G for a number of patients that will rise in the first 4 years post-launch to 125,000 users, and remain at that level for the balance of the period of protection against generic competition.
Including R&D expenditures by the government, Product G will have required about $100 million in uncapitalized expenditures to develop, including Company G spending to complete Phase III trials and secure market approval. The figure of 125,000 daily enrollees is 50 percent of the current number of daily enrollees in treatment for cocaine abuse. The price of $2.50 per daily dose is comparable to the current price of LAAM. The average enrollee takes 12 weeks worth of prescriptions. The company assumes that, given what amounts to 12 years of orphan status and market penetration guaranteed by the government for Product G, no significant competing drug (generic or non-generic) will appear before 13 years post-launch. The company assumes that a competing drug (generic or other) will enter the market at that time, and that this new product will completely overtake Product G within 5 years.
The company does not typically invest in a project unless it has a PAR of at least $300 million and an NPV of at least $100 million. However, its corporate mission does provide for undertaking "public service" projects that might otherwise not meet standard corporate financial goals, as long as such projects present no risk of significant financial loss to the company.
(Note: For the purposes of this scenario, it is not stated how the government has had access to the compound. This may occur, e.g., by the government discovering the compound, by obtaining it from industry or academe, or by working with a compound that is off patent. In this scenario, the patent will expire in 3 years. From the standpoint of Company G, the development work to date has been conducted by another entity at no cost to Company G.)
Base scenario. Under the government's proposed arrangement, the company determines that the PAR of Product G would be $114 million, and that the NPV to the company of investing in Product G would be $142 million, assuming a 12 percent cost of capital.
Base without extended orphan status and guaranteed market. If orphan status remains at the typical duration of 7 years, the guaranteed market runs for 7 (rather than 12) years, and a generic competitor enters the market thereafter, the PAR would remain at $114 million and the NPV would drop to $114 million.
Base without any orphan status and guaranteed market. If no orphan status is accorded, there is no guaranteed market, and generic competition could start as soon as 3 years post-launch and replace Product G in 5 years, the PAR would drop to $91 million and the NPV would drop to $39 million. It is assumed that Product G is still on track to reach peak prescriptions of 125,000 by 4 years post-launch, except that the entry of generic competition 3 years post-launch of Product G precludes reaching that peak, thereby reducing the PAR.
The company notes that the NPV is sensitive to the combination of orphan status and guaranteed market, and that both PAR and NPV are highly sensitive to that combination in the first 7 years.
Pursuant to the provision in its corporate missions for public service, and given assurances of the government provisions for orphan status and guaranteed market, the company decides to take on the project.
Time to patent expiration: 3 yrs.
Time to product launch: 3 yrs.
Premarket R&D expenditures: $100m
Cost of capital: 12%
Orphan drug/similar status: yes
Post-launch to peak prescriptions: 4 yrs.
Peak daily patients: 125,000
Average weeks prescription per year: 13 weeks
Average daily dose wholesale price: $2.50
Time post-launch to competing drug: 13 yrs.
Time for competing drug to replace: 5 yrs.
Scenario 4: Vaccine
In Scenario 4, the government has conducted preclinical R&D on a highly promising Product V that is designed to work as a vaccine with an annual booster.
The government approaches a large pharmaceutical firm, Company V, to take on the project beginning in Phase I trials. The government offers to turn over the rights to the Product V in exchange for conducting all clinical trials, securing FDA approval, and an active marketing campaign through at least 8 years post-launch. The patent will expire in 16 years, about 7 years after expected product launch. Contingent on FDA approval and the marketing campaign, the government will: (a) award orphan drug-like status, (b) add another 3 years of orphan status at the end of the original orphan protection period, and (c) guarantee (with federal and state participation) a wholesale price of $1,000 per patient per year (for the initial vaccine and for subsequent annual boosters) that will rise in the first 5 years post-launch to 500,000 users, and remain at that level for 10-years post-launch.
In order to ensure the considerable market penetration of the drug, the government is committed to expanding treatment and financing capabilities at the state level. This commitment involves the following elements: (a) providing more funding to increase treatment capacity, (b) requiring all substance abuse block grant recipients to offer approved anti-addiction medications, and (c) assuring appropriate financing of new medications by state drug agencies and their counterpart Medicaid agencies.
Including R&D expenditures already made by the government, Product V will require about $200 million in uncapitalized expenditures to develop. The figure of 500,000 peak users is about twice the current number of daily enrollees in treatment for cocaine abuse, and about 25 percent of all heavy cocaine users in the country. The average enrollee will have one treatment, by injection or inhaler, once per year. The price of the treatment, $1,000 per person per year, is equivalent to $2.74 per patient per day for each day of the year.
The company understands that, since 7 years of patent protection will remain after launch, the orphan status will provide R&D tax breaks but no additional protection from generic competition. However, the extra 3 years of extended orphan status will provide another 3 years of protection from generic competition in years 8 through 10 post-launch.
The company assumes that a competing drug (generic or non-generic) will be available on the market following year 10 post-launch. Seven years after its launch, the competing drug will completely replace Product Y on the market.
The company typically does not invest in projects unless they have a NPV of at least $100 million and PAR of at least $300 million.
Under the proposed arrangement, the company determines that that the PAR of Product V would be $499 million, and the NPV would be $254 million.
Company V decides to take on the project.
Time to patent expiration: 16 yrs.
Time to product launch: 9 yrs.
Premarket R&D expenditures: $200m
Cost of capital: 12%
Orphan drug/similar status yes
Post-launch to peak prescriptions: 5 yrs.
Peak daily patients: 500,000
Average weeks prescription per year: 52 weeks
Average daily dose wholesale price: $2.74
Time post-launch to competing drug: 11 yrs.
Time for competing drug to replace: 7 yrs.
Scenario 5: Second Indication
Company S has a smoking cessation medication, Product S, that was recently introduced to the market. In its first two years, Product S has been highly successful, with projected PAR exceeding $500 million. Company researchers also have found strong early evidence that Product S may be very effective as a medication for cocaine addiction, and are encouraging executives to pursue this as a second indication for Product S.
Given previous research and related experience with Product S, the company estimates that it could conduct Phase II and III trials and secure market approval for about $20 million in uncapitalized expenditures. (This is equivalent in the model of taking on the last 7 years of a typical full 13-year product development cycle with total uncapitalized expenditures of $50 million.)
The patent for Product S will expire in 5 years, prior to the anticipated approval of the new indication for cocaine addiction. Company S assumes that if a cocaine addiction indication is approved, Product S will be accorded 7 years of orphan drug-like protection from generic competition at the time of approval. Company S also assumes that immediately following expiration of orphan status, a competing drug will enter the market, and that after another 10 years, it and/or other competing drugs will overtake Product S for the cocaine addiction indication.
The company assumes that the wholesale price Product S will be $3.00 per daily dose, and that it will take 5 years post-launch to reach its target market of 125,000 patients, or 50 percent market penetration of the 250,000 current daily patients enrolled in treatment.
Company S typically seeks new products with PARs of $300 million or more. However, it may have a lower threshold for products with promising second indications that would require relatively lower development costs. Executives are concerned that any modest financial returns from the cocaine addiction market for Product S might be outweighed by any deleterious effects on the current lucrative Product S market of any stigma or adverse clinical events that may arise in association with the use of the product in treatment of cocaine addicts.
Base scenario. Company analysts determine that, for the second indication of cocaine addiction, and given orphan status, the PAR of Product S would be $137 million and the NPV would be $83 million.
Base + achieve $250 million PAR. The company determines that, in order to achieve a more acceptable PAR of $250 million, and still assuming orphan status and peak market of 125,000, the wholesale price per daily dose would have to be $5.50. The company also determines that, in the absence of orphan status, the wholesale price per daily dose would have to be $6.10 to achieve a PAR of $250 million.
Base + low penetration. Noting the relatively low market penetration to date of other substance abuse medications, the company determines that, assuming orphan status and the wholesale price per daily dose of $3.00, a market penetration of 25,000 (10 percent of the current 250,000 daily enrollees) would yield a PAR of $27 million and NPV of $7 million.
Given their knowledge of the market experience of LAAM and naltrexone, company executives are skeptical about reaching the target market of 125,000 with Product S priced above $3.00. As such, they judge it is unlikely that the product could achieve corporate PAR targets. Executives are not favorable toward the possibility of threatening any significant portion of the strong current smoking cessation market of Product S with any stigma or other adverse experience in connection with a substance abuse indication that could yield as little as a $27 million PAR and barely positive NPV. Analysts point out that the patent on Product S will expire before approval for the cocaine abuse indication would occur, and that the strength of the smoking cessation market of Product S could change.
Company S decides not to pursue the second indication for cocaine addiction.
Key Parameters:
Time to patent expiration: 5 yrs.
Time to product launch: 7 yrs.
Premarket R&D expenditures: $50m
Cost of capital: 12%
Orphan drug/like status yes
Post-launch to peak prescriptions: 5 yrs.
Peak daily patients: 125,000
Average weeks prescription per year: 13 weeks
Average daily dose wholesale price: $3.00
Time post-launch to competing drug: 8 yrs.
Time for competing drug to replace: 10 yrs.
Discussion
As illustrated in the "Big Pharm Cold Start" scenario, the prospects of developing a new medication for cocaine abuse and taking it through a full product development cycle do not appear favorable given a moderate wholesale price comparable to LAAM ($2.50 per patient per day) and what amounts to an optimistic target market of 125,000 patients (i.e., 50 percent of the estimated 250,000 people currently enrolled in treatment for cocaine abuse). Although the R&D tax breaks of orphan status can provide modest improvements in NPV, orphan status affords no additional protection for products whose patents have yet to expire. The prospects of delayed competition, in the form of orphan status or delayed entry of a non-generic competitor, can make modest improvements in financial outlook, although its effect on NPV may appear small from the standpoint of a decision maker who is discounting cash flow that will occur 13 or more years in the future.
The Big Pharm scenario does illustrate that, in order to achieve financial indicators that are more in line with traditional targets of large companies, a considerable improvement in price and/or market penetration must be realized. Even assuming the 50 percent penetration of current patients, a wholesale price of $6.60 per patient per day, which is half again as high as naltrexone and two-to-three times the price of LAAM, would be required to achieve a PAR of $300 million. On the other hand, a modest and perhaps more realistic penetration of this market of 12 to 16 percent (i.e., 30,000 to 40,000 patients) could yield a PAR in the neighborhood of $300 million if a cocaine medication were priced at the premium levels that are afforded triple pharmacotherapy for HIV/AIDS, i.e., $10,000 per patient per year (equivalent to $27.40 per patient per day with perfect compliance all year.) More conservative analysis assuming a price equivalent to LAAM ($2.50 per day), a 10 percent penetration of the 250,000 current daily patients, no assurance of orphan status, and the appearance of a competing drug sometime soon after launch would yield a PAR of $20 million and an NPV of a loss of $71 million.
The "Biotech Gets Help" scenario suggests that, even for a company that is confident that it can develop a highly promising molecule with a relatively modest level of R&D expenditures and somewhat lower targets for financial performance, some combination of additional incentives may be needed. In this scenario, three main assumptions are made about government interventions: (a) regulatory reform that would shorten the time to launch by 1 year, (b) provision of market protection similar to orphan drug status, and (c) a significant commitment to expand treatment and financing capabilities at the state level. The expansion of treatment and financing at the state level are assumed to enable the drug to reach a peak market of 250,000 daily enrollees. Given these assumptions and a wholesale price of $2.50 per day, the scenario yields a PAR of $228 million and an NPV of $36 million, both acceptable to the company. Removing each of the government interventions lowers the financial indicators by varying levels. Removing the regulatory reform that shortens time to launch by a year decreases NPV modestly. Removing orphan status also decreases NPV modestly, and does not affect revenue because orphan protection from generic competition would not apply for a drug that will not be off patent. In contrast removal of provisions to expand treatment and financing that could reduce the market size substantially, here down from 250,000 to 125,000 daily enrollees (still an optimistic figure), would reduce PAR by half and push NPV closer to break-even. The drop in these figures would be likely to reduce significantly the number of small companies that would be willing to pursue such a project. Finally, a more pessimistic assumption of market penetration (though realistic in light of the experience of LAAM and naltrexone) of 25,000 (10 percent of current enrollees) would yield unacceptable figures of a PAR of only $23 million with a loss of $18 million for NPV.
In the "Guaranteed Handoff" scenario, the government is offering the rights to a drug that is well along in development (i.e., well into phase III trials) to a company in exchange for the company's finishing the development process and securing market approval. In addition, the government would (a) award orphan drug (or similar) status, (b) provide additional years of market protection from generics, and (c) guarantee purchases for up to 125,000 daily users for the years in which market protection, i.e., (a) and (b), apply. In this scenario, effectively decreasing a company's investment and shortening the time to product launch shifts the risk-reward tradeoff.
Under the government's proposed arrangement, the PAR would be $114 and the NPV would be $142 million. Although the PAR would not be particularly attractive to most large companies under typical circumstances, it may be to smaller companies. In this scenario, where orphan status confers both market protection and R&D tax breaks, removal of the extended orphan status and guaranteed market in the out-years (i.e., after the initial 7 years post-launch) lowers NPV by about 20 percent. Removing the orphan status and guaranteed market in the initial 7 years (in which losses are less cushioned by discounting than in later years) reduced both indicators substantially, i.e., PAR to $91 million and NPV to $39 million. In this instance, PAR is affected directly by the decreased market penetration due to loss of orphan protection from generics, and NPV is afforded less cushioning of revenue decreases by discounting in these early post-launch years. Even so, given the positive NPV, a smaller company might take on the project or, as described in this scenario, a larger company with a corporate mission for public service might still be willing to take on the project.
The "Vaccine" scenario poses more of an outlier set of market conditions involving a promising medication that could be taken just once a year (e.g., vaccine with annual boosters), which may help to obviate compliance problems. As in the "guaranteed handoff" scenario, this involves initial government development of the medication and an offer (earlier in development) to transfer rights to a company to take the product through the balance of development and onto the market. In this scenario, the government provides extended generic protection. Further, the government provides for a substantial, assured market in the form of guaranteed purchases at a premium price ($1,000 per patient per year, equal to $2.74 per day all year) for a number of users rising to 500,000, i.e., twice the current number of daily enrollees in treatment for cocaine abuse, and remaining at that market size through 10 years post-launch. Under these conditions, the PAR would be $500 million and the NPV would be $254 million. This scenario helps to illustrate that extraordinary conditions may be required to bring PAR and NPV over the thresholds sought by the larger pharmaceutical companies.
The "Second Indication" scenario portrays a variation on the risk-reward tradeoff that involves a decision about whether to pursue a market if doing so might jeopardize a currently successful market. In this scenario, a company has a commercially successful product for a CNS indication in a large market that also shows promise for treatment of cocaine addiction. Given previous research and experience with the drug, the company considers that the additional development costs required to secure approval for the second indication would be relatively small. Further, the company expects orphan protection for this indication. Under a base scenario with a moderate price and 50 percent market penetration, the PAR would be $137 million and the NPV would be $83 million. In order to achieve a more palatable PAR of $250 million with the same market penetration, the price would have to be raised to $5.50, which exceeds that of naltrexone. Without orphan status, the price would have to increase to $6.10 to achieve a PAR of $250 million. With more conservative assumptions of a market penetration of 25,000 daily users (10 percent of current daily enrollees) and back to $3.00 per daily dose, the PAR and NPV would drop to $27 million and $7 million, respectively. This scenario illustrates how conservatism regarding expectations for price and market penetration alone can stanch a project. Aversion to the prospects of substance abuse stigma transferring to an already successful product may be secondary, but it could contribute to outweighing any perceived financial returns of a second indication strategy. Under a scenario where the original indication for the product had failed (e.g., if the drug had not reached the market or had been a commercial failure), there may be less down-side risk of pursuing a cocaine abuse indication; however, the challenges to price and market size would remain.
Case Studies: LAAM, Naltrexone, Clozapine, and Nicorette
The purpose of the case studies is to gain insight into the experiences of companies that are relevant to developing and marketing medications for drug abuse and addiction. Presented below are two in-depth case studies of LAAM (Roxanne), for heroin addiction, and naltrexone (DuPont Merck), for heroin (as Trexan) and alcohol (as ReVia) addiction. In addition, we present two smaller case studies on clozapine (Sandoz), for schizophrenia, and Nicorette (SmithKline Beecham), for smoking addiction. These case studies address the following general topic areas: (1) product history and development timeline; (2) clinical development and product positioning issues; (3) product marketing strategy and sales; (4) policy interaction in product development and distribution (e.g., with HHS, FDA, NIH, and DEA); and (5) likely future of the product.
Summary of Case Study Results
The four case studies have several elements in common, particularly with regard to the target patient population. For example, three of the four drugs involve treatment for substance abuse including LAAM and naltrexone for heroin addiction, naltrexone for alcoholism, and Nicorette for smoking. Clozapine was included in this study because the market for antipsychotic drugs shares certain characteristics with the market for pharmacotherapies for drug addiction including: 1) small market size, 2) treatment funding primarily through public sources, 3) and some patients who need help caring for themselves and complying with medication. The four patient populations represented in the case studies serve as highly relevant examples of some of the barriers that may exist in the development of pharmacotherapies for cocaine addiction.
Although each population shares several important characteristics, the condition of each population is viewed differently by outside populations. Differences in the markets for these drugs include the levels of federal funding provided for clinical trials, addictive properties of the drug, and treatment delivery system. As a result of these differences, each case study provides key lessons about the barriers to the development of pharmacotherapies (Figure 22 below).
| Case Study Drug | Key Market Lesson |
|---|---|
| LAAM | Existing delivery system via methadone maintenance clinics created significant market barriers. |
| Naltrexone | Despite excellent pharmacological properties, poor patient compliance and failure to gain acceptance by providers and payors severely limited market penetration. |
| Clozapine | High cost of treatment due to required weekly patient monitoring severely limited market penetration. |
| Nicorette | Fewest distribution barriers and over-the-counter approval boosted sales and led to an influx of competing products. |
As mentioned above, the government played a key role in the development of three of the four case study drugs by lowering some of the market barriers, particularly by funding clinical trials. These strategies are summarized in Figure 23 (below), and more detail is provided below.
Existing Government Strategies to Lower Market Barriers
The government was involved in aspects of development and marketing for each of the four case study drugs. These phases, adapted from the 1995 IOM report, include discovery, clinical studies, NDA phase, and marketing.
| Strategy | Case Study Drugs | ||||
|---|---|---|---|---|---|
| Naltrexone | |||||
| LAAM | Trexan | ReVia | Clozapine | Nicorette | |
| Discovery | |||||
| Funding of basic science research | |||||
| Clinical Studies | |||||
| Funding of clinical trials | |||||
| NDA Phase | |||||
| Fast-track approval | |||||
| Orphan drug status | |||||
| Other market exclusivity | |||||
| Less stringent phase IV clinical trial requirements | |||||
| Marketing | |||||
| Mandated coverage | |||||
| Source: The Lewin Group | |||||
NDA Phase
A) Fast-track approval
The FDA can grant fast-track approval to those drugs that it deems will provide a new therapeutic effect for a particular patient population. For example, clozapine was given a "1A" approval rating because it was hailed as the first break-through antipsychotic drug in 30 years. Similar fast-track approvals were given informally to LAAM and Nicorette. Those drugs given a "1A" approval level are accorded a shorter FDA review time, which lowers the time-to-market barrier and allows a company to start marketing its drug faster than drugs with lower ratings.
B) Orphan Drug Status
All of the drugs in these case studies were granted some form of market exclusivity post-FDA approval, either via orphan drug status or other exclusivity incentives. For example, LAAM was granted orphan drug status primarily because the size of the potential product market, i.e., based on the expectation that users of LAAM would be drawn from the patient population then taking methadone, fell below a U.S. prevalence of 200,000 patients. Orphan drug status can provide 7 years of post-approval market exclusivity as well as tax credits and federal grants for clinical research for the treatment of rare conditions. Orphan drug status serves to lower the investment barrier, while raising the expected returns, thus providing a more favorable NPV than a drug without orphan status.
C) Market exclusivity other than Orphan Drug Status
Naltrexone (as ReVia), clozapine, and Nicorette were given varying lengths of post-approval market exclusivity. Although not as comprehensive as orphan drug status, marketing exclusivity allows a pharmaceutical company to sell its drug for a certain length of time free of competition from generic versions of the drug. This type of marketing exclusivity is often granted to encourage pharmaceutical companies to develop an indication for a drug, e.g., naltrexone, whose patent has expired or to encourage a company to develop an already approved drug for a new indication. With market exclusivity, the expected returns are higher, thus improving the NPV, making entry into the market more appealing.
Pharmacotherapies that are unable to qualify for orphan drug status (e.g., Nicorette) may also apply for market exclusivity under the Waxman/Hatch regulations in the Drug Price Competition and Patent Term Restoration Act of 1984. A new product must meet the following criteria in order to qualify for exclusivity:
- FDA must not have approved an identical drug product with same conditions of use prior to submission of the product's NDA;
- NDA or supplemental new drug application (SNDA) must contain reports on new clinical investigations of the medication, not studies relied on for the approval of another drug product or for a previous NDA;
- Application must be supported by reports on clinical investigations other than bioavailability studies;
- These studies must be essential to approval of the supplement and must be conducted or sponsored by person submitting the supplement (Wright 1997; Tan Sheet, Apr. 15 and Nov. 11, 1996).
D) Less stringent phase IV clinical trial requirements In the case of ReVia, the FDA modified regulatory requirements to encourage DuPont to submit a SNDA for alcoholism. The FDA linked phase IV clinical trials requirements to the annual sales of ReVia. No phase IV trials were required if sales of ReVia did not meet certain thresholds. If ReVia did well on the market, DuPont would have to conduct phase IV trials based on the level of sales. By allowing for flexible phase IV studies, the federal government lowered post-marketing costs, improved NPV projections, and made investment in the alcoholism indication more promising.
Marketing Mandated Coverage
The federal government exercised a coverage mandate in the case of clozapine. Once Sandoz separated its expensive Clozaril Patient Monitoring System (CPMS) from sales of clozapine, the Health Care Financing Administration(now known as Centers for Medicare and Medicaid Services(CMS)) (HCFA(now known as CMS)) required all state Medicaid agencies to cover the cost of clozapine therapy and provide a patient monitoring system of the providers choosing. This helped to increase patient access to clozapine and increase sales. By mandating coverage of treatment for certain conditions, the federal government can ensure that patients have access to appropriate therapies and provide incentives for pharmaceutical companies to pursue a wider market. This can lower the market barrier associated with an uncertain payment structure and result in a more favorable NPV.
Case Study 1: LAAM
Introduction
Drug Overview
LAAM (levo-alpha-acetylmethadol) is a synthetic opioid analgesic marketed under the trade name Orlaam by Roxane Laboratories for the treatment of opioid dependence. The clinical effects of LAAM, comparable to the effects of methadone, allow the medication to serve as a substitute for other opiates (e.g., heroin), suppressing craving and staving off withdrawal symptoms in opiate-dependent individuals. In contrast to methadone, LAAM is able to suppress withdrawal symptoms for forty-eight to seventy-two hours; therefore, the medication is only administered three times per week rather than daily. LAAM may only be administered at federally and state-approved opioid treatment programs (OTPs). In these programs, two treatment approaches are typically used: a) short term treatment (six months or less), or b) long term treatment (lifelong maintenance). Federal regulations require the treatment programs to provide "a comprehensive range of medical and rehabilitative services...that include medical evaluations, counseling, rehabilitative and other social programs...which will help the patient to become a productive member of society" (21 CFR 291).
Market Overview
In 1993, it was estimated that approximately 0.9% of young adults in this country had tried heroin and a significant percentage became dependent on the opiate (Johnston 1994). According to Abt Associates, in 1995, approximately 500,000 people were addicted to opiates. In the United States, there are approximately 1,000 FDA-approved opioid treatment programs (750 methadone maintenance programs and 250 - 300 inpatient hospital detoxification programs). SAMHSA reports that approximately 115,000 (25%) opiate addicts receive treatment from the maintenance programs.
Key Issues from the Case Study
LAAM had an extensive research cycle that lasted over twenty-five years with limited private investments and two unsuccessful New Drug Applications (NDAs). After finally obtaining FDA approval in 1993, the medication confronted severe market barriers largely pertaining to public policy (e.g., regulations on controlled substances, take home medications, reimbursement, treatment of pregnant women) and treatment issues (e.g., inertia of methadone providers, patient preference).
A few key points emerged repeatedly during the telephone interviews for this case study. Certainly, some private respondents believe that government agencies should continue to play an active role in the development of pharmacotherapies for substance abuse addiction. Indeed, LAAM may never have made it to the market without the government's participation, particularly in support of clinical development of the drug. However, given a choice between government funding for R&D with restrictions on marketing versus full control over their product, pharmaceutical companies would choose the latter.
Second, multiple sets of restrictions and regulations, which govern the distribution of the medication in opiate treatment programs, continue to be major barriers to the distribution of LAAM. These include:
- Federal rescheduling of the medication;
- Federal regulations for the OTPs;
- State rescheduling of the medication;
- State regulations for the OTPs; and
- Reimbursement from state and private payers.
Respondents suggested that efforts should be made to streamline these barriers and thereby restore some of the marketing window and increase availability of medications. Alternatively, one respondent recommended maintaining the existing regulations and barriers but extending the period of market exclusivity through legislation (e.g., the Orphan Drug Act).
Third, respondents emphasized that the market for such medications as LAAM differs significantly from the market for traditional pharmaceutical products. Unlike the marketing of traditional pharmaceutical products, pharmaceutical companies marketing substance abuse medications face stringent regulations on distribution, delivery systems, and provider reimbursement. In the substance abuse treatment market, pharmaceutical companies cannot market to pharmacies or physicians; their medications are not prescribed by physicians or obtained over-the-counter. Pharmaceutical companies must market their medication to the opiate treatment programs and clinics throughout the country. These OTP clinics diagnose the patients, develop treatment plans, and administer the medications to the patients. The qualifications of the clinic personnel vary significantly from state to state; in certain cases, clinic managers have limited medical background and limited knowledge in pharmacotherapy. Therefore, pharmaceutical companies report having a difficult time introducing a new medication into these clinics.
Product History and Development Timeline
As shown in Figure 24 (below), LAAM was first developed as an analgesic in 1948. Fraser and Isbell (NIDA 1994) conducted the first clinical study of LAAM in 1952; their research demonstrated that LAAM had the capacity to suppress opiate withdrawal symptoms for over 72 hours. In 1968, Dr. Jerome Jaffe discovered the potential utility of LAAM in opiate maintenance treatment and initiated the clinical research on LAAM. The initial IND application for clinical studies on LAAM was submitted in 1969 by Dr. Jaffe. Clinical research on LAAM, funded mainly by the government, continued throughout the 1970s by various researchers. In 1979 and 1981, NDAs for LAAM were submitted to the FDA by NIDA contractors; however, these NDAs were not approved. On January 24, 1984, LAAM received orphan drug status for the treatment of opiate addiction. By receiving orphan drug status, the medication was granted a 7-year period of market exclusivity following FDA approval. Rosina Dixon, who was the sponsor of LAAM when it obtained its orphan drug status, conducted research on the drug for a period of time but never submitted an NDA. In the mid-1980s, only limited clinical research was conducted on LAAM. Clinical trials and research on LAAM resumed in late 1980s and early 1990s under the sponsorship of NIDA via a contract with Biometrics Research Institute (BRI) to prepare the NDA. In 1993, the FDA approved LAAM for marketing and the seven years of market exclusivity granted under the Orphan Drug Act began; the market exclusivity ends in July 2000.
Figure 24: Timeline for the Development and Approval of LAAM
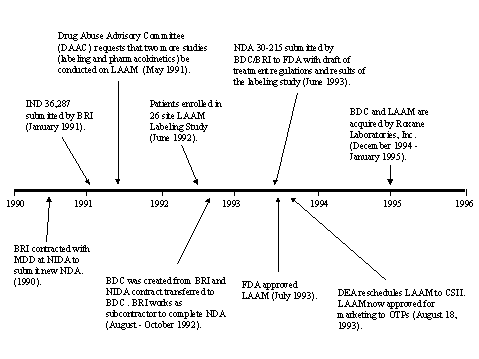
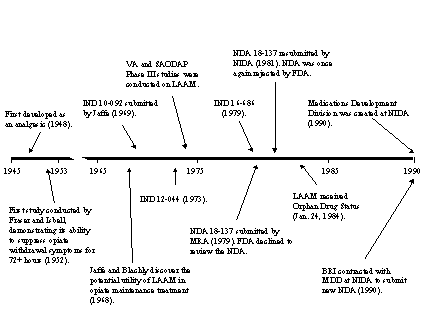
Clinical Development and Product Positioning
Clinical Trials, Phases I - III
As discussed earlier, Fraser and Isbell conducted the initial clinical trials on LAAM at the Addiction Research Center in Lexington, Kentucky. These studies examined the clinical effects of LAAM in "post-addict" subjects and in subjects who were dependent on significant doses of morphine. In addition, these Phase I studies demonstrated that following parenteral administration, withdrawal symptoms were suppressed for 48 hours. The Phase II clinical studies were conducted in the late 1960s and 1970s by Irwin et al. and Blachly et al. (NIDA 1994). These studies focused on the dose response, clinical pharmacology, and safety of the medication. Research from these studies demonstrated that, in contrast to daily administration of methadone, LAAM was to be administered every 48 to 72 hours.
In the late 1960s and early 1970s, the federal government funded two major Phase III clinical studies of LAAM under the sponsorship of the Veterans Administration and the White House's Special Action Office for Drug Abuse Prevention (SAODAP). These two clinical studies were conducted to determine the efficacy and safety of the medication in a treatment setting. Concurrently, Jonathan Whysner of MRA, Inc., was also conducting Phase III clinical trials under an Investigational New Drug application (IND) in 1973 in preparation for the submission of his NDA for LAAM (NIDA 1994).
Initial IND and NDA Applications
During the 1970s, NIDA assumed responsibility for LAAM and contracted with two companies to submit the NDAs. In 1979, the initial NDA was submitted by NIDA in conjunction with Jonathan Whysner of MRA, Inc. The FDA declined to review this NDA based on insufficient documentation in the Chemistry and Manufacturing sections. After the FDA declined to review the application, NIDA purchased it from MRA and revised the application for resubmission. The NDA was resubmitted to the FDA in 1981 by NIDA; once again, this request was denied due to inadequacies in the application (Medications Development Division 1997).
Funding cuts in NIDA research budgets in the early 1980s led to a hiatus in research on LAAM. However, with the advent of the HIV/AIDS epidemic and the realization that needle sharing was placing addicts at high risk for contracting disease, NIDA once again focused their energies and funding on researching LAAM. In 1990, the Medications Development Division (MDD) was created at NIDA. This division became responsible for LAAM and sent out a request for proposal (RFP) for the preparation and submission of an NDA. The RFP received responses from seven different companies including Biometrics Research Institute (BRI); in 1990, NIDA contracted with BRI for $3 million to develop and submit the NDA.
Contract between NIDA and BRI
According to respondents, BRI initially became involved in the development of pharmacotherapies for the treatment of substance abuse addiction in the 1970s. BRI had a contract with NIDA to assist with the collection of data on naltrexone; it worked with NIDA on the medication up to the Phase III clinical trials at which point DuPont became the primary party responsible for naltrexone. Concurrent with BRI's involvement in naltrexone, one of its patients was conducting research on LAAM. After BRI ended its contract with NIDA for naltrexone, it assisted its client, Jonathan Whysner, with the research and development of LAAM.
After receiving NIDA's RFP for the submission of the LAAM NDA, BRI conducted an initial market analysis that examined the number of clinics and patients. Their calculations estimated the percentage of patients that were suitable for LAAM and the percentage of those that would switch from methadone to LAAM. According to officials at BRI, the theory in the 1970s and 1980s was as follows: (i) patients off the street would receive methadone for a period of time; (ii) after the patients stabilized on methadone, patients would then begin receiving LAAM in the clinical setting; (iii) once the danger of relapse was minimized, patients would receive naltrexone and progress to becoming drug-free (Bradford 1997).
Under BRI's contract with NIDA for the submission of the NDA, BRI was responsible for collecting data from past research, conducting any necessary new research, and preparing and submitting the NDA. Government funds ($3 million) were to cover costs for collecting research on LAAM and conducting any new studies. BRI would fund the manufacturing and marketing costs of the medication.
In 1991, after gathering the past data, BRI submitted an IND Application to the FDA. Upon review of the IND, the FDA requested that two additional studies, a pharmacokinetic study and a labeling study, be conducted prior to submission of an NDA. In June 1992, the Labeling Assessment Study (the final clinical study prior to FDA approval) was initiated.
The NIDA contract required BRI to market and distribute the medication upon submission of a successful NDA. BRI's greatest strengths were in clinical trials and data management associated with NDA development and not in product marketing and distribution. Therefore, BRI elected not to engage in the marketing of the drug. In August 1992, officials from BRI left the company in order to create BioDevelopment Corporation (BDC) for the purpose of marketing and distributing LAAM. BDC was created by three principals who raised the funding for their company through private channels. After its creation, BDC assumed the contract from NIDA. BRI remained a subcontractor and continued to assist with the preparation of the NDA. In June 1993, BDC submitted an NDA for LAAM to the FDA; this application was approved by the FDA in July 1993, eighteen days after submission. After the medication received FDA approval, the DEA and FDA worked quickly to reschedule LAAM. In August 1993, one month later, the FDA and DEA agreed upon the rescheduling of LAAM to a controlled substance II (CSII) with a "no take-home" policy. FDA incorporated this policy into the labeling of the medication, a labeling that also restricted the use of the medication by women of childbearing age. After obtaining FDA and DEA approval, BDC was now permitted to market the medication to the opioid treatment programs.
Product Marketing Strategy and Sales
Before BRI responded to NIDA's RFP, BRI conducted a market analysis to ascertain the potential market size and the number of potential patients for LAAM in this market. Prior to FDA approval, BDC conducted its own market analyses to project its return on investment. According to their calculations, 715 public methadone maintenance clinics were in operation and serving approximately 90,000 - 110,000 patients daily. Officials at BDC hypothesized that 50 to 60 percent of the current patients were possibly suitable for treatment with LAAM. BDC believed that the advent of LAAM could potentially reduce the costs of treatment by decreasing the number of patient visits per week and thereby decreasing administrative costs. Officials at BDC therefore believed that the clinics would now be able to serve 10,000 - 20,000 new patients on the same budget for treatment services (Bradford 1997).
However, during the market analysis in 1993, BDC and BRI did not fully account for the existing regulations and restrictions that had to be met prior to medication distribution. BDC assumed that virtually no state or local regulatory barriers existed. For example, BDC assumed that once the DEA had rescheduled the medication, states would quickly follow suit and reschedule LAAM. However, by August 1993, BDC began to perceive the barriers that had to be overcome prior to distribution of LAAM.
Barriers to Distribution
In contrast to traditional pharmaceutical products, LAAM had to overcome many barriers after FDA approval. To permit the distribution of LAAM to treatment programs, states first had to reschedule the medication from a Schedule I drug (permitted only for clinical research) to a Schedule II drug (permitted for restricted use in treatment settings). Certain states, such as Texas, have "automatic rescheduling" where medications rescheduled by DEA are immediately rescheduled by the states 30 days after the DEA decision. Other states, such as Michigan, have pharmacy boards that convene periodically to review the rescheduling of controlled substances. Finally, in some states such as California and Florida, the legislature must pass legislation authorizing the rescheduling of medication. Such legislative efforts may require significant time and may require the drafting and enactment of legislation, delaying market entry in states by months and/or years (NIDA 1994).
Even after rescheduling the medications, many states had to revise or amend existing regulations for OTPs. The OTPs must adhere to strict guidelines established by the federal, state, and, in some instances, county governments. In many states, the restrictions were specifically directed toward "methadone maintenance programs." Therefore, to allow clinics that administer LAAM to operate legally, states had to revise the laws so that the regulations applied generally to opioid treatment programs rather than to a particular type of treatment program (e.g., methadone maintenance).
Following each state's own approval process for the medication, BDC had to focus its energies on the issues of reimbursement for the medication. Many OTPs received funding for their clinics from state general appropriations. The funds were directed toward covering the operating costs of the OTPs and for the reimbursement of the administration of the medication. Costs of the medication were and continue to influence clinics' decisions to distribute LAAM. According to the 1996 Drug Topics Red Book, methadone dosage costs approximately $0.50 per day while LAAM costs approximately $2.00 per day (accounting for LAAM being taken three times per week). According to data collected by Capital Consulting Corporation, methadone purchasing costs per patient per day ranged from $0.32 to $0.55 compared to LAAM costs that ranged from $0.71 to $1.53 per day. In certain states, Medicaid would reimburse clinics for the cost of methadone but would not cover the cost of LAAM. Therefore, clinics either had to negotiate with the state to receive greater funding or the clinics had to absorb the additional costs associated with LAAM. This uncertainty regarding medication reimbursement and state funding as well as costs have served as major barriers to the distribution of LAAM in many clinics.
Another barrier to the distribution of LAAM at the clinic level (besides costs and reimbursement) may be the inertia of the methadone providers. Respondents acknowledged that the personnel in many clinics seemed resistant to change and to the implementation of a new medication. The introduction of a new medication in the clinical setting demands new protocols, new training, and new reimbursement mechanisms and negotiated rates. Staff acceptance and their positive support for the medication are an important step toward patient acceptance. Prior to staff acceptance, personnel need to undergo training to understand the medication and its differences from methadone. Once the barriers at the state and clinic levels are overcome, the medication may become an integral component of treatment in clinics.
Patient acceptance of the medication is integral to the distribution of the medication in the clinics. Initially, patients had strong negative perceptions about LAAM and had heard rumors about the medication including the following:
- Women were concerned that LAAM would impact their menstrual cycle.
- Men were worried that LAAM would reduce their sex drive.
- Patients associated LAAM with cancer, liver problems, nervousness and other major medical difficulties.
- Patients recalled hearing of problems with LAAM from 1970s clinical research, in which some patients were thought to have died from the medication (Feldman 1994).
In the past three years, patient perceptions of LAAM have changed significantly. Many patients report that they feel "more normal" on LAAM than they have in a long time, including their time on methadone. Despite increased patient support for LAAM, patients have also voiced their opposition to LAAM's "no take-home policy" which is inconvenient and a marked difference from methadone. Take home methadone may be given only to a patient who, in the reasonable clinical judgment of a program physician, is responsible in handling narcotic drugs. In determining whether a patient is responsible for handling narcotic drugs, a physician may consider the following: absence of recent abuse of drugs, regularity in clinical attendance, absence of serious behavioral problems, and length of time in maintenance treatment. According to the FDA regulations, a patient who has satisfactorily adhered to the program rules for at least 3 months and who has "made substantial progress in rehabilitation" may receive no more than a 2-day take-home supply of medication. A patient who has adhered to the program rules for at least 2 years may receive no more than a 3-day take-home supply of medication. Clinics throughout the country vary in the stringency of their take-home policy.
Marketing Strategies
One marketing strategy employed by BDC was to capitalize on LAAM's infrequent administration and the subsequent decreased administrative costs, relative to methadone. BDC marketed this advantage to the staff of OTPs, expecting a positive response. However, staff and clinic personnel became upset, fearing that their funding could be cut with the advent of LAAM; clinics also believed that states would compel the clinics to treat a greater number of patients with the same funding.
Realizing the negative response to this marketing approach, BDC sought to highlight LAAM as an alternative to methadone. Methadone must be administered daily and patients have to take methadone home if clinics were not operating over weekends. In contrast, LAAM only requires administration three times per week and operates under a "no take-home" policy. Patients do not need to interrupt their daily schedule in order to receive medication; communities do not need to be concerned with the diversion of LAAM on the streets. However, emphasizing the strengths of LAAM over methadone as a marketing strategy did not seem to improve the market penetration of the medication. While a "no take-home policy" was a positive attribute of LAAM in the eyes of people in the communities, clinics often viewed the "no take-home" policy of LAAM as a disadvantage. The following example illustrates the complex market interactions at the local level.
In one small Western city, LAAM was introduced into one of the city's two private methadone clinics. The availability of LAAM produced a significant increase in patients (and revenue) for this clinic (clinic A), since patients were attracted to the reduced visit schedule offered by LAAM. The city's other clinic (clinic B) responded to the loss of patients (and revenue) by liberalizing its take home methadone policy, so that patients would be less attracted to the availability of LAAM in clinic A. The liberalization of the take home policy was such a successful marketing strategy that a dramatic increase in the enrollment in the clinic B resulted. Many of the clinic A patients transferred to clinic B. Clinic A responded to this loss of business by increasing its availability of take home methadone. According to[a] survey respondent in clinic A, "with LAAM the only positive is a reduced visit schedule. With take home methadone, patients get a reduced visit schedule and some extra income (i.e., illicit take home sales) to help pay for clinic fees." (Rawson 1996)
As highlighted by this change of events, it was not profitable for clinic A to administer LAAM given the resulting activity of clinic B, namely the liberalized take-home methadone policy. On the other hand, creating an OTP that administers LAAM in a city with no treatment programs may be profitable for a clinic. However, distributing LAAM in one clinic in a city with other OTPs may generate a loss for the LAAM clinic.
Market Revenues
As discussed above, BDC did not anticipate the barriers to market penetration following FDA and DEA approval. Prior to the creation of BDC, its principals raised $2 million to cover the initial start-up costs. An additional $2 million was raised during the first 12 to 18 months of the company. According to officials at BDC, it was anticipated that the start-up funding would need to last for 18 to 24 months before the sales of LAAM would reach "break-even" and begin to generate a profit. BDC had estimated that approximately 20,000 patients would be receiving LAAM at 24 months (Bradford 1997).
Figure 25 (below) highlights the difference between the projected number of patients receiving LAAM and the actual number of patients over the last four years. As demonstrated by the following graph, the sales from LAAM fell well short of BDC's expectations and its threshold values. After 24 months, only approximately 2,000 patients were taking LAAM, significantly lower than the projected 20,000 patients. After 48 months, it is estimated that approximately 5,000 patients (less than 5 percent of persons in OTPs) are receiving LAAM daily (Roxane and BDC estimates).
Figure 25: Expected and Actual Number of Patients Receiving LAAM
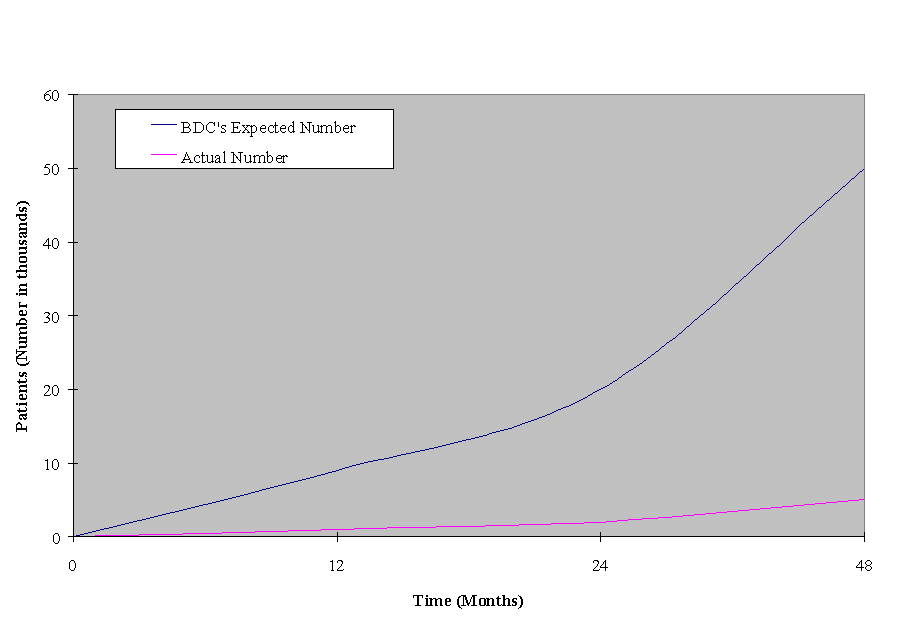
Source: Industry estimates.
Acquisition of LAAM by Roxane Laboratories
After 12 months in the market, BDC realized that its funding was insufficient to support the continued marketing and distribution of LAAM. In the fall of 1994, BDC began looking for a pharmaceutical company with which to merge. Offers were received from three small firms and two larger pharmaceutical companies, Roxane Laboratories and Mallinkrodt. Roxane had already established itself in the field of substance abuse with the acquisition of methadone from Lilly. BDC decided to join Roxane in part due to this experience. In January 1995, Roxane Laboratories acquired BDC and the marketing and distribution rights to LAAM.
Prior to acquiring BDC, Roxane conducted its own internal market analysis. The focus of the market analysis was not the overall potential magnitude of the market but rather the percentage of the potential market that could be realized and the time required to realize that market. In an effort to further market penetration of LAAM, Roxane has conducted extensive training of clinic staff to facilitate the introduction of LAAM into the clinical setting. Staffs have been educated on the positive attributes of LAAM and have heard testimonials from patients already receiving the medication.
The penetration of LAAM has increased slightly since Roxane's acquisition of BDC, and Roxane Laboratories holds the medication in high regard and will continue to distribute the medication to clinics throughout the country.
Experience of LAAM in the VA clinics
In contrast to other public clinics, the VA has been successful at distributing LAAM. It is estimated that approximately 10% of the patients taking LAAM are receiving the medication in clinics funded by the VA. According to Dr. Richard Suchinsky, Associate Chief of Addictive Disorders at the VA, the hospitals have introduced the medication into all 33 OTPs within their systems and approximately 10 - 15% of their patients are receiving LAAM. Certain VA clinics in the larger cities are even directly inducting patients onto LAAM rather than transferring patients from methadone to LAAM. The successful distribution of the medication in the VA system may be attributable to three different factors. First, the VA clinics have a strong medical presence with physicians who understand the pharmacokinetics of the medication. Second, the VA clinics have not had to confront the same number and level of regulations and restrictions as other public OTPs. Third, in contrast to the state and local OTPs, the VA clinics have not had significant difficulties obtaining reimbursement for the medication. Finally, the VA hospitals were involved in the LAAM Labeling Assessment Study conducted in 1993 prior to FDA approval.
Policy Interaction in Product Development and Distribution
The interaction of policy with LAAM product development and distribution occurred throughout the thirty year period.
Government Involvement in Clinical Development
The federal government's role as a major financier of research on LAAM during the 1970s was instrumental in promoting earlier clinical trials. The Phase III studies of note funded by the government, the VA and SAODAP studies, were conducted to determine the safety and efficacy of the medication. The government was also instrumental in reinvigorating the research on LAAM in the late 1980s. NIDA funded the Labeling Assessment Study and contracted with BRI (and subsequently with BDC) to submit the NDA for LAAM that was subsequently approved by the FDA in July 1993.
Government Involvement in Product Marketing and Distribution
While government involvement during clinical trials was supportive, government involvement following FDA approval was more restrictive. As discussed above, many of the market barriers confronted by the medication pertained to federal and state policies (e.g., regulations on controlled substances, reimbursement issues, and take-home restrictions). Prior to FDA approval, the government had granted LAAM orphan drug status that gave the medication 7 year market exclusivity after FDA approval; the market exclusivity does not conclude until July 2000. However, the government restrictions and market barriers have significantly shortened this period by increasing the time required for the product to penetrate the market.
Marketing to Public Clinics
The marketing of LAAM has been directed exclusively to state approved public programs and to programs within the VA. The pharmaceutical companies have not directed their marketing energies toward private clinics because there is little data available on the number of private clinics and the number of patients receiving treatment. According to BDC, it was difficult to ascertain the size of the market but BDC believed that the market was not substantial. Due to the absence of concrete data and statistics, BDC and Roxane have focused their time on the public and VA clinics.
Likely Future of Product
At this point in time, LAAM has captured less than five percent (approximately 5,000) of the patients in the OTPs. An additional barrier to market penetration is the existing cap on the number of patients that may be treated in the individual OTPs. To establish an opioid treatment program, the clinic must obtain approval from the state authority and local government planning boards. In approving these clinics, these officials often examine the proximity of other clinics and the size of the treatment population. Depending on these and other circumstances, clinics may have a difficult time obtaining approval from the state or the local boards. The budgets for the OTPs have effectively established a de facto cap on the number of patients that may be treated in OTPs. Therefore, to further LAAM's market penetration, patients would have to be shifted from methadone to LAAM (CSAT 1994).
Clinical Trials and New Indications
Currently, Reckitt & Colman Pharmaceuticals, Inc. (in collaboration with NIDA), is developing buprenorphine and buprenorphine combined with naloxone as treatments for opiate dependence. Once approved, these products will compete with methadone and LAAM in the existing system, challenging methadone and LAAM's current market share.
In addition to the clinical trials being conducted on a new medication for opiate addiction, academic-based research on LAAM is continuing. Dr. Walter Ling of the Friends Research Institute and the University of California at Los Angeles (UCLA) is conducting a study on the efficacy and safety of take-home LAAM; the results of this study may be utilized in an effort to change the labeling concerning LAAM's "no take-home" policy. In addition, Dr. Doug Anglin at UCLA is conducting research comparing the effectiveness of methadone to LAAM in reducing of HIV transmission (Medications Development Division 1997). At this point in time, however, Roxane Laboratories is neither conducting nor sponsoring any research on take-home LAAM or the possible effects of LAAM on women of child-bearing age.
The Behavioral Pharmacology Research Unit at The Johns Hopkins University recently completed a study on the dose-related efficacy of LAAM for opioid dependence. According to the researchers, "the results indicate that LAAM's efficacy as an opioid dependence pharmacotherapy is related to dose and that high-dose LAAM is safe and efficacious for male and female drug abusing patients." These results support earlier data regarding the safety of the medication for women of child-bearing age, an issue addressed in LAAM's labeling (Eissenberg 1997).
Conclusion
Since its approval by FDA in 1993, LAAM has captured less than five percent of the patients in OTPs. Despite its ability to suppress withdrawal symptoms longer than methadone and despite its ability to make the patients feel "more normal," LAAM has only penetrated the market to a limited extent for various reasons. According to respondents from the private sector, it has been difficult to introduce LAAM into the market in part because methadone has existed as the sole medication for opiate addiction for over 25 years. Also, it has been difficult to break the virtual monopoly that methadone has established in the market, especially given the higher cost of LAAM. Aside from methadone's hold on the market, the main barriers to the marketing and distribution of LAAM have been the following:
- Rescheduling of LAAM to a Controlled Substance II by state agencies. The process for rescheduling the medication varies significantly from state to state. Certain states automatically rescheduled the medication after the DEA while other states have required legislation to approve LAAM. This process has often taken prolonged periods of time. Indeed, three states have yet to approve the medication for their OTPs.
- Amendments to state licensing regulations for opioid treatment programs. Many states had regulations that applied to "methadone maintenance programs". To authorize the use of LAAM in licensed clinics, those regulations needed to be amended to incorporate all opioid treatment programs.
- Reimbursement for LAAM by states. Securing reimbursement for the medication has been especially difficult because on average, LAAM costs more than methadone.
- Staff resistance to change. Staff at clinics have resisted the medication in part due to their negative perceptions of it. In addition, the introduction of a new medication requires new training and new protocols, and raises potential resource constraints on many clinics that are already underfunded.
- Patient resistance to the "no take-home policy" on LAAM. While patients have stated that LAAM makes them feel "more normal," the no take home-policy still serves as a major deterrent to LAAM given the availability of take-home methadone.
These barriers have served as formidable obstacles for market penetration and have significantly reduced the value of the 7 year market exclusivity period of LAAM extended by its orphan drug status. According to respondents from the private sector, the existence of these barriers and the relatively small size of the potential market will continue to deter pharmaceutical companies from investing funds in the R&D and marketing of pharmacotherapies for the treatment of opiate addiction.
Case Study 2: Naltrexone
Introduction
Drug Overview
Naltrexone is a pure opioid antagonist originally marketed by DuPont as Trexan for the treatment of opioid dependence, and later as ReVia for the treatment of alcohol dependence. Naltrexone binds competitively to opioid receptors in the brain, and thus blocks the physiologically reinforcing euphoric effect of exogenous opioids like heroin. Naltrexone also plays a role in blocking the endogenous opioids associated with alcohol consumption, thereby blocking the euphoric effects of alcohol. Naltrexone works to help addicts control their craving for either heroin or alcohol by eliminating the euphoric effects. Naltrexone is typically distributed through treatment centers as part of comprehensive treatment programs for the treatment of opioid and alcohol dependence.
Market Overview
Estimates of the number of opioid addicts (primarily heroin) in the U.S. range from 500,000 to 1,000,000 (IOM 1990, Abt Associates 1995, Hammil and Cooley 1990). These estimates have remained fairly stable since the 1970s. In contrast, 15.3 million people in the U.S. are afflicted by alcohol abuse and dependence (Pink Sheet 1995, Scrip 1993). There were approximately 650,000 patients in alcohol treatment centers in 1992 (NDATUS 1993).
Key Issues from the Case Study
Naltrexone provides two related yet distinct pharmaceutical R&D and marketing lessons. Naltrexone's development encompasses over 30 years and reveals a wide range of government involvement in the drug development process, from conducting and funding clinical trials to creating novel regulatory approval incentives. For example, the impetus and vast majority of funding for clinical development came from the federal government through the National Institute on Drug Abuse (NIDA) and the National Institute on Alcohol Abuse and Alcoholism (NIAAA). Interviewees emphasized that the development of naltrexone required a true public-private collaboration between the federal government and DuPont, the company that owned the rights to naltrexone. However, the story of naltrexone also reveals that despite significant government efforts and a willingness of DuPont to pursue naltrexone's development, a number of market barriers have prevented naltrexone from becoming successful. To date, sales of naltrexone for both the heroin treatment indication and the alcohol treatment indication have fallen far short of DuPont's original, modest expectations. In both cases, federal government support of naltrexone's clinical development was necessary but not sufficient to overcome some of the key market barriers.
A major market barrier with naltrexone is low patient compliance. There are several reasons patient compliance on naltrexone therapy is low, and there are several consequences. There is a large barrier to the initiation of naltrexone therapy because patients must be completely opioid free. Many addicts return to heroin use before detoxification because they are not able to cope with the physiological withdrawal effects of complete detoxification. State-level treatment centers for heroin addicts have not been able to afford the more intensive psychosocial support systems necessary to ensure patient compliance to Trexan. In addition, according to our interviewees, directors of federally funded clinics are often non-clinicians who do not understand how a non-addictive alternative to methadone can be effective for heroin treatment despite evidence from clinical trials which demonstrated that naltrexone was highly effective if taken. Low patient compliance also limited the marketing of naltrexone, as ReVia, to comprehensive alcohol treatment centers.
The following sections provide a more comprehensive overview of the research, development, and marketing experiences of naltrexone.
Product History and Development Timeline
Executive and legislative mandates of the 1970s provided the impetus for the development of naltrexone as a narcotic antagonist to treat the rapidly rising number of heroin addicts, both in the U.S. and in U.S. military personnel abroad. The timeline for the development of Trexan is presented in Figure 26 (below). The timeline for ReVia continues in Figure 27 (below). In June 1971, President Nixon created the Special Action Office for Drug Abuse Prevention (SAODAP), which consolidated all federal agencies that had resources devoted to drug abuse and addiction research. The SAODAP was first directed by Dr. Jerome Jaffe. By September, 1971, the Division of Narcotic Addiction and Drug Abuse (DNADA) of the National Institute of Mental Health (NIMH), in conjunction with SAODAP, had initiated a research plan to expedite the development of a narcotic antagonist, that could be used as a non-addicting pharmacotherapy for the treatment of opioid addiction. In March 1972, Congress passed the Drug Abuse Office and Treatment Act, with a particular interest in developing "long-lasting, non-addictive, blocking and antagonist drugs or other pharmacological substances for the treatment of heroin addiction." This Act provided financial support for research in this area. In 1973, DNDA separated from NIMH and became the National Institute on Drug Abuse (NIDA) (Julius 1976).
Figure 26: Timeline for the Development of Naltrexone as Trexan
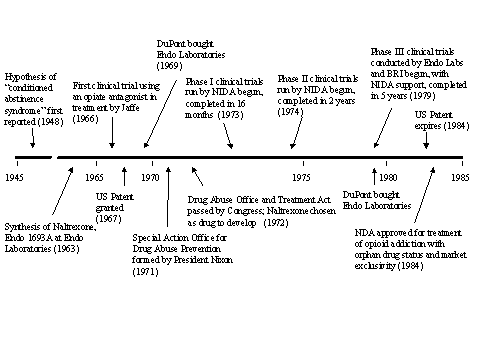
Source: The Lewin Group
SAODAP and DNADA selected naltrexone for further development because, of the other drugs in early development at the time, naltrexone came the closest to meeting their 12 criteria for an ideal narcotic antagonist:
- ability to antagonize the euphoric high of opiates
- absent or low-agonistic effects, especially unpleasant ones
- does not cause physical dependence
- does not exhibit increasing tolerance to its antagonistic actions
- absence of serious side effects and toxicity even in chronic use
- easily administered, i.e., no surgery or painful procedure involved
- long-lasting or moderate duration of antagonist effects
- absent or low abuse potential
- reversible effects in case of medical emergency
- high potency to allow administration of small amounts in a biodegradable vehicle
- easily available and inexpensive
- therapeutic efficacy in treatment of narcotic addiction (Julius 1976).
SAODAP rejected several drugs in favor of naltrexone. For example, SAODAP rejected naloxone because of its high cost, difficult synthesis, poor oral absorption rate, and short duration of action (Julius 1976). It rejected cyclazocine because of its potential for strong agonist properties as well as other drugs that were considered to be too early in development.
Naltrexone was originally synthesized in 1963 and patented in 1967 as Endo 1639A (U.S. patent number 3332950) by Endo Laboratories, a small pharmaceutical company in Long Island, New York. Endo was a manufacturer of pain relief medications and had conducted substantial research on narcotic agents. Naltrexone was a cyclopropylmethyl analog of naloxone (used as an antidote for acute opiate overdose). In 1969, DuPont purchased Endo Laboratories to acquire a company with pharmaceutical marketing experience to help sell DuPont's Symmetrel, an antiviral compound. With the purchase of Endo, DuPont acquired the rights to several successful drugs, including: Coumadin (warfarin), an anticoagulant; Percodan, a prescription narcotic; Nubain, a combination agonist/antagonist analgesic; and Naloxone, for narcotic overdose. DuPont acquired the rights to naltrexone, still in the early development phase, as part of the overall purchase of Endo.
In 1972, SOADAP approached DuPont for permission to develop naltrexone for clinical use. At the time, it seemed unlikely that DuPont would develop naltrexone. First, DuPont thought naltrexone would have relatively low market potential. Second, the patent for naltrexone would most likely have expired before clinical development would have been completed. Third, thebaine, a chemical precursor to naltrexone, would have to be purchased from Mallinkrodt, the pharmaceutical company that isolated thebaine.
Ultimately, NIDA asked for DuPont's assistance in facilitating naltrexone's transit through the FDA regulatory process - in particular to identify the required clinical trials and to file the NDA. DuPont interviewees reported that a primary reason for helping the government to bring naltrexone to the market was the company's "public spirited" mission. DuPont agreed to assist NIDA with the development of naltrexone, particularly with filing the NDA, regardless of the economic returns. In return, NIDA agreed to pay for the bulk of clinical development costs.
The clinical trials for naltrexone as a treatment for heroin addiction began in 1973 (Schecter 1974, O'Brien 1978). The NDA for heroin treatment was approved in 1984, the same year the U.S. patent expired. On March 11, 1985, naltrexone was designated an orphan drug which provided 7 years of post-approval market exclusivity.
The impetus for the funding of clinical research to gain an alcohol indication for naltrexone also came from the federal government. Researchers at VA hospitals who had been using Trexan to treat heroin addicts noticed that treatment with Trexan reduced both heroin and alcohol use (O'Brien 1996, Volpicelli 1995, and O'Malley 1995).
Figure 27: Timeline for the Development of Naltrexone as ReVia
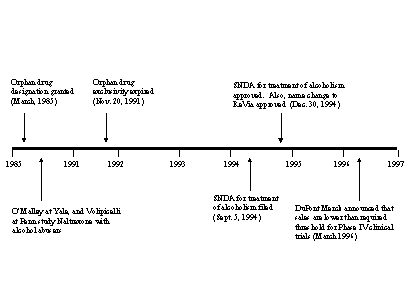
Source: The Lewin Group
The funding for the development of the alcohol indication for naltrexone was provided by the NIAAA. The SNDA for the alcohol treatment indication was approved in 1994, providing 3 additional years of market exclusivity for the alcohol indication (Pink Sheet 1994).
Clinical Development and Product Positioning Issues
A large amount of preclinical research on opioid receptors and narcotic antagonists had been underway prior to the development of naltrexone (Crabtree 1984), but naltrexone was the first narcotic antagonist to be clinically tested and developed for the treatment of heroin addiction (Schecter 1980).
Clinical Trial Results
The clinical trials showed a modest success in the reduction of heroin use. Factors that made treatment successful included: sustained therapy with naltrexone, participation in multidisciplinary programs, and good family and social support (Crabtree 1984). The early clinical trial results showed that compared with the methadone maintenance patients, those patients who were attracted to naltrexone therapy were relatively "more motivated and emotionally stable" (Schecter 1974). In 1974, Schecter and colleagues found that naltrexone successfully blocked the pharmacologic effects of heroin (Schecter 1974). Clinical trials by Martin et al. (1973) and Resnick et al. (1974) and the National Research Council Committee on Clinical Evaluation of Narcotic Antagonists (1978) showed that although naltrexone was an effective opiate blockade, clinical success, i.e., a reduction in heroin use, was limited to fully compliant patients. Similar results were found in clinical trials for alcoholism (O'Brien 1996). As a result of these findings and others, the labeling for naltrexone reads, "[Naltrexone], unlike methadone or LAAM (levo-alpha-acetylmethadol), does not reinforce medication compliance and is expected to have a therapeutic effect only when given under external conditions that support continued use of the medication" (naltrexone package insert).
Clinical Trials Obstacles
One unanticipated obstacle during the clinical development of naltrexone was the difficulty in patient accrual and compliance for the clinical trials, which resulted in much higher costs than initially anticipated. Dr. Arnold J. Schecter, who conducted many of the early safety and efficacy studies for naltrexone in the treatment of opioid addiction at the State University of New York, reported that patient recruitment was difficult because some patients feared a new drug, lacked a desire to become drug free, were unwilling to possibly receive a placebo, and disliked the rigid protocols associated with the clinical trials (Schecter 1980). In addition, patients had to remain opiate free for a minimum of 5 to 10 days prior to treatment because naltrexone would cause severe withdrawal symptoms in patients with opioids in their system (Schecter 1974). Many addicts were unable to remain opioid-free for the required amount of time because of the physiological withdrawal effects. Finally, unlike methadone treatment which helps to suppress craving, naltrexone had no effect until the addict attempted to use heroin. Some patients feared that when on naltrexone they would be more vulnerable to these heroin cravings and felt that methadone was more effective in controlling their cravings.
Many researchers also encountered suspicion in the community regarding treatment of patients with a new experimental drug. The methadone maintenance clinics were especially reluctant to refer patients for naltrexone therapy, partially because of their need to keep their own censuses high enough to receive funding (Schecter 1980). As a result of these difficulties recruiting patients, the naltrexone clinical researchers made no efforts to screen out patients who can be difficult to manage in clinical trials, e.g., patients who were poorly compliant, and this may have compromised the results of the clinical trials (Schecter 1980).
The methadone maintenance clinics felt that naltrexone therapy was less effective and more costly than methadone for two primary reasons. One reason was that heroin addicts would have to be completely opioid free prior to starting naltrexone therapy. This meant that, unlike methadone maintenance, heroin addicts undergoing naltrexone treatment would experience all of the physiological symptoms of opioid withdrawal creating a huge hurdle for initial compliance to therapy. Second, naltrexone therapy required more extensive psychosocial support services than methadone treatment, primarily because naltrexone was non-addictive and lacked the reinforcing effect of methadone. Schecter and colleagues (1974) estimated that total clinical treatment with naltrexone was almost twice as expensive as methadone treatment (an increase from methadone's annual per patient cost of $1200 - $1700 to a cost of $3500 per year) because of this need for more intensive psychosocial services. Naltrexone supporters argued that naltrexone therapy would be substantially more economical when compared with inpatient beds, jail facilities or other therapeutic communities (Schecter 1974). In addition to the difficulties of patient compliance, Dr. Schecter noted that the lack of adequate funding of the antagonist clinics, well below funding of methadone clinics, was another factor in the limited success of the early clinical trials (Schecter 1980).
Naltrexone had a few additional clinical side-effects or problems that concerned many treatment providers. First, naltrexone did not prevent addicts from using other drugs to experience a euphoric effect. Second, there was a danger of opioid overdose in those patients who tried to overcome the naltrexone blockade. Third, patients on naltrexone would have to use pain medications that did not rely on opiate action, and patients were encouraged to carry a card that indicated they were on naltrexone in the event of an emergency. Finally, some practitioners feared an increased chance of depression, although this was not clinically verified (Schecter 1980). These problems compounded the problem of low patient compliance and created significant barriers to the clinical acceptance of Trexan.
The clinical trials for alcohol treatment encountered similar problems with low patient compliance. Naltrexone did not perform significantly better than a placebo unless it was administered as part of a comprehensive, multidisciplinary treatment program (O'Malley 1995). As a result the labeling for ReVia included the following stipulation, "ReVia should be considered as only one of many factors determining the success of treatment of alcoholism." This labeling indication had a profound effect on product marketing strategy and sales by limiting marketing to comprehensive alcohol treatment programs.
Naltrexone researchers for both opioid and alcohol indications faced many barriers during the course of their research including difficulties with patient recruitment, patient compliance, the high cost of clinical support services, and the traditionally low funding of treatment centers. While the government funded and supported the clinical trials, the funding fell short of the amount necessary to provide the more intensive psychosocial support. Researchers also faced difficulties recruiting patients, which meant that all patients who agreed to participate in the clinical trials were accepted into the treatment program. The researchers did not "reject" any patients from the clinical trials. This may have negatively affected the results of the clinical trials by including a high proportion of high-risk patients, who may have been motivated more by payments for participating in the trial than addiction treatment which lead to poorer compliance and higher drop-out rates (Schecter 1980). These barriers had a significant impact on DuPont's marketing efforts after Trexan and ReVia were approved.
Product Marketing Strategy and Sales
DuPont did not expect Trexan or ReVia to become major revenue generators. Just prior to the launch of ReVia, Trexan sales were approximately $5-8 million annually, which represented approximately 15-25,000 patients per year, or less than 5% of the estimated number of heroin addicts (Scrip 1993). Trexan was marketed only through comprehensive treatment centers at a price of $3.80 per patient day (Scrip 1993). When Trexan's name was changed to ReVia and the alcohol treatment indication was added, DuPont expected U.S. sales of ReVia to rise to $15-25 million annually, which represented approximately 45-80,000 patients per year, or 1% of alcohol addicts and opioid addicts in the U.S. (Scrip 1993). For the treatment of alcoholism, the recommended dosage of ReVia is one 50mg tablet per day for up to 12 weeks. When it came on the market in 1995, ReVia was priced the same as Trexan at wholesale prices of $227.58 for 50 tablets at 50mg (Pink Sheet 1995).
As of October, 1996, DuPont Merck reported that sales of ReVia since market entry in January 1995 had been lower than expected. As of October, 1996, DuPont had not yet reached the FDA's threshold of 200,000 prescriptions that would have required them to conduct phase IV clinical trials (Pink Sheet 1996). The primary market barriers of patient compliance and the need for more intensive and expensive psychosocial support than other existing therapies significantly limit market penetration. Trexan has failed to penetrate the highly regulated federal treatment market for opioid addiction, and ReVia has failed to gain coverage under most private insurance plans. For both indications, DuPont sales representatives perceived that they had to manage provider and patient expectations by reminding them that naltrexone was not a "cure" for addiction. DuPont also had to convince providers that it was appropriate to treat a drug addiction with a pharmacotherapy. The marketing of naltrexone remains subject to significant barriers, and DuPont has not been successful in selling ReVia except in limited cases. (For example, the VA hospital system has widely adopted the use of naltrexone in the treatment of alcoholics.) These market barriers remain the most persistent and the most difficult to overcome.
Methadone maintenance clinics for the treatment of heroin addiction are subsidized by the federal government and highly regulated at both the federal and state levels, in part because methadone is a controlled substance. Naltrexone is not a controlled substance and does not fall under the same regulatory umbrella as methadone. However, funding for the treatment of heroin addicts was funneled primarily through methadone maintenance clinics at the state level. Thus DuPont's marketing strategy for Trexan focused on working with the methadone clinics, which were primarily controlled by state health care agencies such as the State of New York Division of Substance Abuse. Representatives from DuPont noted they also marketed to private hospitals or "white collar" treatment areas because patients in private hospitals tended to be more highly motivated and have a stronger support network, and would experience more favorable treatment outcomes.
DuPont's Marketing Strategy
DuPont had an extremely difficult time trying to convince methadone clinic personnel to use naltrexone once it was approved. One barrier was that clinics would have to implement more intensive psychosocial support programs to promote patient compliance. Most facilities could not afford to implement naltrexone therapy due to the combined price of the drug, the drug treatment program, and the additional time for counseling.
DuPont did not launch a targeted marketing campaign to physicians, primarily because non-physician administrators often made the crucial funding and regulatory decisions that affect the care given in the facility. DuPont marketed naltrexone as a non-addictive antagonist blocking therapy that would help rid patients of opioids completely. DuPont also stressed that the patients would have to be motivated to comply with the therapy. The DuPont sales force had a difficult time explaining the antagonistic mechanism of naltrexone and its benefits to a lay audience that was uninformed about the science underlying naltrexone and the drug's mechanism of action. The sales force reported that they were entering a consumer marketplace with inherent misunderstandings and negative perceptions. Pro-methadone treatment providers argued that because methadone was dependence-producing, it was easier to maintain a patient on methadone and thus more likely that treatment would be successful. One former member of the DuPont sales force said these misunderstandings continue to be a great barrier to the use of naltrexone.
After working around or within the methadone treatment camps, DuPont gave up trying to convert proponents of the "methadone philosophy" to the benefits of treatment with antagonists. These two treatment camps were very strongly divided. DuPont found a favorable audience in the private heroin treatment clinics. Specifically, the clinicians in the private clinics reportedly had a better understanding of the clinical benefits of naltrexone therapy. Also, private clinics could more easily afford the additional psychosocial therapy to help maintain patient compliance because some private insurers covered naltrexone treatment.
The publicity and stigma surrounding treatment of substance abuse was another market barrier for DuPont to negotiate. There was a negative public perception of methadone clinics as a "taxpayer-supported program that keeps junkies addicted." As a non-addictive blocking agent, naltrexone was perceived much more favorably than methadone. However, the favorable view of naltrexone raised expectations to the extent that naltrexone was being touted as the "cure of opioid addiction." Clinical trials results showed that naltrexone would not cure the addiction, but naltrexone would enhance the chances of a successful recovery if used as part of a comprehensive treatment process. Favorable expectations raised initially by the press could not be met. DuPont salesmen devoted considerable effort to managing these expectations and explaining the importance of naltrexone therapy in conjunction with a comprehensive treatment program.
Marketing Strategies for ReVia
The alcohol treatment market is very different than the opioid market. At the time ReVia entered the market, there were a few potential competitors with products that all demonstrated poor clinical results. For example, the market for disulfiram (marketed as Antabuse) was limited in its clinical effectiveness because of poor patient compliance. Other treatments that had been tried, including off-label use of antidepressants like fluoxetine, demonstrated poor results in treating alcohol abuse. ReVia was a significant improvement over disulfiram in its safety profile and potential for improved patient compliance and outcomes. In addition, because the treatment system was not as highly regulated as the heroin treatment system, DuPont had more flexibility in marketing directly to the clinics and treatment providers. Despite ReVia's clinical superiority over disulfiram and less restrictive distribution channels than for heroin treatment, DuPont's sales force encountered similar marketing problems.
Clinical trials with alcohol showed similar results to the clinical trials with opioids. In an outpatient setting, ReVia, administered via a prescription to the patient, was not much more effective than a placebo. However, in comprehensive alcohol treatment programs, ReVia was very successful in helping patients reduce their alcohol consumption (O'Malley 1995). Therefore, DuPont marketed naltrexone only through comprehensive inpatient and outpatient alcohol treatment centers, including the VA hospital system where the clinical trials were conducted.
While DuPont could have marketed its product directly to general practitioners, there were many reasons why it did not. DuPont did not want naltrexone to be falsely construed as a "miracle pill" that would "cure alcoholism," because it could not stop all alcoholics from drinking, especially without counseling from comprehensive treatment centers. Additionally, DuPont stressed that even a reduction in alcohol use was a desirable outcome (Behavioral Health Treatment 1996). As with Trexan, there is a strong camp of treatment providers who feel that alcoholism should not be treated by substituting one drug for another and that alcoholics were not cured unless they were abstinent. For these reasons, DuPont wanted to ensure that ReVia was marketed as a complementary, rather than stand-alone, therapy.
Interviewees reported that another barrier to market penetration is that ReVia has been unsuccessful in gaining formulary access with private insurance companies. For example, a chain of California treatment centers using naltrexone as the primary pharmacologic treatment suspended operations after only six months citing managed care companies' lack of desire to cover such treatment (Behavioral Health Treatment 1996). Managed care companies may be reluctant to cover naltrexone treatment because few cost-effectiveness studies on treatment with naltrexone have been done. However, employers are also responsible for limiting substance abuse treatment coverage in their employee's health plans (Buck 1997).
Policy Interaction in Product Development and Distribution
In addition to conducting and funding most of the clinical trials for Trexan and ReVia, the federal government implemented several policies to further promote the development and distribution of naltrexone.
The FDA gave Trexan orphan drug status, granting DuPont seven years of post-approval market exclusivity. When ReVia was approved, the FDA granted DuPont three additional years of market exclusivity for the alcoholism indication. These market exclusivity rulings protected DuPont from generic versions of the medication.
The FDA also added a novel step-wise regulatory incentive for phase IV clinical trials for ReVia. The FDA allowed DuPont to tailor the type of phase IV studies that had to be conducted based on the extent of use of the product, in a four-tiered system based on the number of annual prescriptions. If the number of prescriptions stayed below 200,000 per year, DuPont would be exempted from any phase IV requirements. If the number of prescriptions rose above 200,000 per year in the first three years, DuPont would conduct pharmacokinetic studies in patients with severe renal disease and severe hepatic disease. At each of 500,000 and 1,000,000 prescriptions per year, DuPont would conduct a series of additional phase IV trials including studies in older adults and children, patients with common co-morbid conditions, and larger patient populations. DuPont is also required to provide in its annual report to the FDA the amount of drug manufactured as well as the number of prescriptions (new or refill) filled annually based on IMS data. This ruling allowed the FDA to make the product available to a small number of patients without requiring DuPont to conduct expensive phase IV trials, while also ensuring that additional data would be collected if the product was used more widely (Pink Sheet 1995). (Note: DuPont and Merck & Co. formed a partnership in 1991 known as DuPont Merck, which owns the rights to Trexan and ReVia. DuPont Merck markets its products under the DuPont Pharma name.)
Likely Future of the Product
DuPont has essentially stopped actively marketing both Trexan and ReVia. The company still provides information to clinicians upon request and makes naltrexone available to clinicians for use with their patients on an as needed basis. Researchers have approached DuPont for permission to test naltrexone for use in a wide variety of other conditions including obesity, schizophrenia, and chronic obstructive pulmonary disease (Watson 1996). The FDA awarded orphan grants (research grants for treatments of rare conditions) for the use of naltrexone with a number of rare conditions, including childhood autism (FDA 1989). In addition, researchers have used derivatives of naltrexone in other conditions. For example, the FDA granted orphan drug status to methyl-naltrexone as a drug that blocks the side effects of morphine without interfering with pain relief in cancer treatment (Oncology 1996). However, because naltrexone is now completely off-patent, DuPont will most likely not pursue any other indication for naltrexone without a guarantee of market exclusivity post-approval.
Conclusion
Naltrexone held great clinical promise when it became an important figure in the federal government's efforts to combat heroin addiction in the 1970's. As a non-addicting antagonist of the euphoric high of opiates, naltrexone had certain characteristics of a drug that promised to revolutionize the treatment of heroin addiction. Naltrexone showed great promise in clinical trials by significantly reducing heroin use in patients. The federal government made the development of naltrexone a top priority, and created consolidated government divisions with the funding necessary to conduct the clinical research and development.
Orphan drug status and related provisions for market exclusivity, and the flexible phase IV trial requirements are all useful tools employed by the FDA to give DuPont more incentive to develop naltrexone. However, other barriers described above overshadowed the advantages gained from these regulatory incentives.
The federal government played a key role in eliminating many barriers to the development of naltrexone for both heroin and alcohol addiction. Several key barriers the government recognized and lowered, and their effect on DuPont's incentive to market the drug, are shown in Figure 28 (below).
| Market Barrier | Government Intervention | Effect |
|---|---|---|
| High expense of clinical development, with a low projected return | The federal government funded and conducted the vast majority of clinical trials | Lowered DuPont's initial development costs and made the initial research less risky financially |
| Difficulty of obtaining thebaine, a precursor to naltrexone | The NIH negotiated with Mallinkrodt to obtain thebaine | DuPont had easier access to an essential precursor material |
| Patent expiration | FDA granted orphan drug status and additional market exclusivity | "Protected" the market to increase DuPont's chances of gaining a return on their investment |
| High expense of post-marketing studies | FDA granted variable Phase IV research requirements | DuPont could bring the drug to the market without having to conduct costly Phase IV trials until the drug was widely used |
Despite the high level of government intervention, there were several barriers that DuPont and the federal government were unable to overcome. Estimates of naltrexone's sales suggest that naltrexone has reached less than 5% of heroin or alcohol addicts. As of October, 1996, sales of naltrexone had not reached the FDA's minimum threshold of 200,000 prescriptions per year to require phase IV research. As shown in Figure 29 (below), many of these barriers are related to issues of patient compliance.
| Market Barrier | Effect on DuPont's Marketing Success |
|---|---|
| Patient compliance | The non-addictive property of naltrexone, which was a high priority in choosing to develop naltrexone, makes patient compliance difficult. Clinical trials had shown that naltrexone was not effective without additional psychosocial therapy. |
| Need for intensive psychosocial therapy in addition to drug treatment | Limits the distribution of naltrexone through comprehensive patient treatment centers, and potentially misses another market segment that may seek care primarily through a general practitioner. |
| Higher cost for more intensive psychosocial therapy | State level treatment centers can not afford to implement the more intensive psychosocial support systems necessary to maintain patient compliance on naltrexone. In addition, there was no established reimbursement system for naltrexone treatment at the state level. |
| Federally controlled heroin treatment system | As a result of extensive regulations at the federal and state levels, DuPont had to market directly to individual state substance abuse directors, who often lacked clinical backgrounds. |
| Difficulties recruiting patients for clinical trials | The methadone maintenance clinics were reluctant to allow their patients to enter clinical trials for naltrexone either because they needed to protect their patient censuses, or they believed that a non-addictive alternative to methadone would not be effective. |
| Unrealistic patient and provider expectations | Naltrexone was not a "miracle pill" that would cure a patient of all addictions and DuPont sales representatives had to manage provider expectations and convince them that reducing consumption of opioids or alcohol was significant. |
| Lack of insurance coverage | Although some heroin addicts are covered under Medicaid, many private insurers are unwilling to cover naltrexone. In addition, managed care companies and employers offer limited insurance coverage for substance abuse. |
| Provider reluctance to use pharmacotherapies in the treatment of substance abuse | Many providers in the addiction world share a philosophy that substance abuse should not be treated with drugs, which further impeded DuPont's sales efforts. |
Case Study 3: Clozapine
Introduction
Drug Overview
Clozapine is a tetracyclic dibenzodiazepine antipsychotic agent marketed by Sandoz Pharmaceuticals under the trade name Clozaril in the U.S. and Leponex in the rest of the world. It is part of a class of drugs known as "atypical antipsychotic agents" which do not exhibit the typical extrapyramidal side-effects of antipsychotic agents including acute dystonia, rigidity, tremor, and akathisia. Clozapine has been used to treat schizophrenia, nonschizophrenic psychotic states, depression, neuroses, and behavioral disorders (Ereshefsky 1989). Clozapine has demonstrated remarkable results in treating patients with schizophrenia, particularly those who were previously treatment-resistant. Unfortunately, clozapine has been associated with an elevated rate (2% of all patients taking clozapine) of agranulocytosis characterized by a sharp decline in white blood cells making the patient more susceptible to potentially fatal infections. As a result, the FDA required that a comprehensive case-management system be used by all providers who dispense clozapine to ensure that patients' white blood cell counts are monitored weekly. The use of clozapine was also restricted to three sub-populations: (1) treatment-resistant patients with schizophrenia (approximately 10-20% of patients with schizophrenia), (2) patients who cannot tolerate the extrapyramidal symptoms of conventional anti-psychotics, and (3) patients with evident tardive dyskinesia that is not suppressed. (Tardive dyskinesia is characterized by involuntary repetitive movements of the facial, buccal, oral, and cervical musculature. Unlike most anti-psychotics that cause tardive dyskinesia in 15-20% of patients, clozapine has not been found to cause tardive dyskinesia.) (Ereshefsky 1989). Clozapine therapy must be initiated in an inpatient setting, to ensure the safety of the patient by titrating the dosages to reduce the risk of agranulocytosis. As described in this case study, the risk of agranulocytosis and the need for stringent and expensive patient monitoring was a major barrier to market penetration.
Market Overview
Approximately 1% of Americans (2.4 million people) are afflicted with schizophrenia, and between 10 - 25% (approximately 250,000) of these patients receive little or no benefit from conventional anti-psychotics (Cruzan 1989). Sandoz estimated that of these, approximately 33% (60-80,000) would respond to clozapine therapy (Pink Sheet 1991). Although patients with schizophrenia represent only a small portion of the U.S. population, they account for almost 25% of all inpatient beds used for any medical treatment in the U.S. The schizophrenia pharmacotherapy market is estimated at $1.1 billion annually and includes haloperidol, the most widely prescribed antipsychotic drug, clozapine, risperidone (approved in 1993), olanzapine, perphenazine, among others (FDC Reports 1997).
In addition to having a relatively small patient population, the market for drugs for schizophrenia shares a number of other characteristics with the substance abuse population. For example, both markets share Medicaid as a primary payer, a state-run treatment system, and a patient population that often has trouble with activities of daily living. For the schizophrenia market, approximately 90% of schizophrenia patients in the U.S. are Medicaid recipients, many are treated in state-run hospitals, and patients with schizophrenia are more likely to be non-compliant with pharmacotherapy than patients without schizophrenia (In the case of schizophrenia, non-compliance is attributed to the fact that patients with schizophrenia do not understand their condition). The similarities of the markets make the inclusion of a case study on a treatment for schizophrenia especially pertinent to the development of pharmacotherapies for substance abuse.
Key Issues from the Case Study
As much of the initial research and clinical development of clozapine was conducted outside the U.S. and funded by Sandoz, the clinical development issues are less relevant to U.S. R&D efforts. Rather, clozapine's post-regulatory and marketing periods are more relevant segments of this case study, in light of the FDA's novel regulatory requirement of a patient monitoring system and prohibitively high cost of clozapine treatment which severely impeded sales despite high clinical demand. These issues were resolved only through legal action involving the Food and Drug Administration (FDA), the Federal Trade Commission (FTC), the Health Care Financing Administration(now known as Centers for Medicare and Medicaid Services(CMS)) (HCFA(now known as CMS)), and providers such as the Department of Veterans Affairs (VA) Hospitals who demanded a lower cost of clozapine distribution.
The development of clozapine relates to the development of pharmacotherapies for substance abuse because a private pharmaceutical company invested its own money to develop a drug despite a small market. The fact that Sandoz marketed clozapine to a worldwide market helped to lower the barrier of small market size. However, the drug faced a significant market penetration barrier in the U.S. and Canada when sales of the drug were linked to an expensive patient monitoring system. Once distribution of the drug was separated from the patient monitoring system, sales of clozapine in the U.S. doubled. This case study provides an example of how a tightly controlled drug delivery system and a high price can impede market penetration.
Product History and Development Timeline
Clozapine was developed in Eastern Europe by the Wander division of Sandoz Pharmaceuticals. Clozapine's clinical development can be divided into essentially two phases, or "attempts." The first "attempt," depicted in Figure 30, began with clozapine's discovery and synthesis in 1952, and ended in 1975, when clozapine was withdrawn from worldwide markets after an outbreak of clozapine-associated agranulocytosis.
Clozapine was first patented in the late 1950s in Switzerland. European clinical trials began in 1962. Clozapine was first patented in the U.S. on November 10, 1970. U.S. clinical trials began in 1972, 10 years later after the European trials. From 1973 until 1975, clozapine was marketed for the treatment of schizophrenia in 22 countries (in Europe, Asia and Africa) under the trade name Leponex.
Figure 30: Timeline for Clozapine Development, Part I
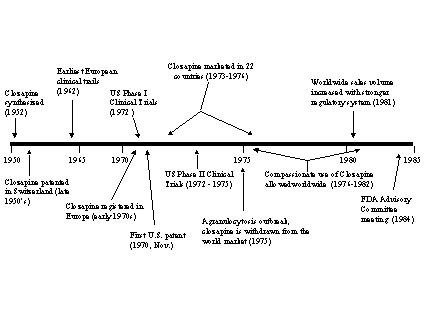
Figure 31: Timeline for Clozapine Development, Part II
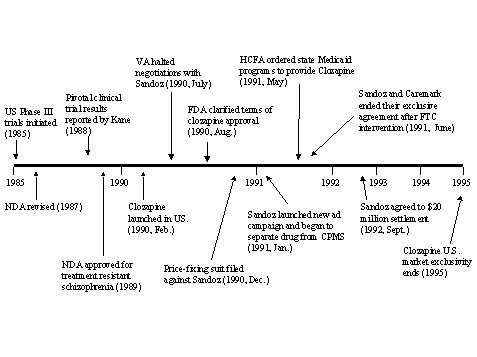
In 1975, clozapine was withdrawn from the world market after 16 cases of clozapine-associated agranulocytosis resulted in 8 fatalities in Finland (Anderman 1977). Other cases of clozapine-associated agranulocytosis and fatalities were reported to a lesser extent in other countries. Compassionate use of clozapine was allowed in research settings from 1976 to 1982. In 1981, as providers developed stricter patient monitoring systems, the sales volume of clozapine began to increase.
The second "attempt" at the clinical development of clozapine began in 1984, after an FDA advisory committee approved further U.S. clinical development of clozapine, as shown in Figure 31 (above).
U.S. phase III clinical trials recommenced in 1985 and the U.S. NDA was approved in 1989. The FDA approval stipulated that Clozaril (U.S. trade name) be sold only with a patient monitoring system to prevent fatalities due to agranulocytosis. In February 1990, Sandoz introduced Clozaril in conjunction with the Clozaril Patient Management System (CPMS) to promote the monitoring of patients' white blood cells to identify and control cases of agranulocytosis. Pricing of Clozaril in connection with the CPMS triggered marketing resistance to the drug, as described below. In December 1990, 33 states and the District of Columbia filed suit against Sandoz claiming that the company was "price-fixing" by limiting the sale of a highly desired product for a limited patient population through a monitoring system that was prohibitively expensive, and which could be provided by health care providers less expensively.
The remainder of this case study will discuss in detail the events after clozapine entered the U.S. market, and how the resulting bundling issues were resolved.
Clinical Development and Product Positioning Issues
The product history and clinical development of clozapine is not directly pertinent to the case study, as most of the clinical development occurred in Europe. There were very few barriers to diminish the intent of Sandoz to develop their antipsychotic drug. From initial clinical trials, the drug demonstrated improved efficacy over competing products without the same extent of extrapyramidal side-effects. The main barriers faced by Sandoz arose from marketing issues in the U.S. associated with the CPMS.
Product Marketing Strategy and Sales
When clozapine was approved by the FDA in 1989, the agency required that the drug be distributed with a patient monitoring system. The FDA had several reasons to require a patient monitoring system including: (1) a high incidence of a potentially fatal adverse reactions, (2) the inability to identify in advance patients who will suffer the reaction, (3) the probability that the risk of death can be substantially reduced by weekly testing and monitoring, (4) FDA's experience that physicians do not always comply with label recommendations for relatively burdensome testing and monitoring, and (5) an unusual patient population that cannot be relied on to take responsibility for regular testing (Peck 1990).
Sandoz implemented a novel marketing tactic of selling clozapine exclusively through the CPMS in conjunction with Caremark, a division of Baxter, and Roche Biomedical Laboratories, which tied distribution of the drug to weekly white blood cell monitoring. The drug and the CPMS were bundled at a price of $172 dollars per week, which resulted in a cost of approximately $9,000 (1990 dollars) per patient per year making clozapine therapy approximately 8 to 15 times more expensive than therapy with traditional anti-psychotics (Tokarski 1990). Clozapine was sold primarily to state mental health hospitals, the VA hospitals, and state Medicaid programs. Sandoz was criticized for charging so much for a drug that was desperately needed for a relatively small group of patients whose treatment was funded almost entirely through taxpayer dollars, via Medicaid.
Sandoz had several reasons to justify the high cost of the drug in conjunction with the CPMS, including: (1) to recover research and development costs, (2) to cover the costs of the monitoring system, (3) to ensure financial stability with the U.S. patent for clozapine set to expire only 4 years after market approval, and (4) to reduce the chances that Sandoz would be subjected to liability suits due to cases of agranulocytosis resulting from improper monitoring.
This marketing strategy created a significant price barrier to rapid diffusion of the product. By July 1990, only 5 months after clozapine was introduced in the U.S., the VA hospitals stopped providing clozapine to patients because of the restrictive cost (Tokarski 1990). By December 1990, almost 1 year after clozapine was introduced, only 7,100 patients (of a target market of 250,000) were taking clozapine. The poor distribution of a needed drug and provider lawsuits prompted the federal government, through legal and regulatory activity, to force Sandoz to separate the drug from the patient management system.
As shown in the graph below, in less than one year after Sandoz separated the drug from the patient management system and HCFA(now known as CMS) mandated that clozapine be included on state Medicaid formularies, sales of clozapine in the U.S. doubled. Sales of clozapine have been increasing since 1991, despite increasing competition. In 1993, clozapine enjoyed a worldwide sales growth of 60%. In 1993, Janssen's risperidone (Risperdal) entered the market as the second atypical antipsychotic, creating stiff competition for clozapine. In 1996, clozapine's worldwide growth was 17% with 1.7 million worldwide prescription sales and a 7% share (based on number of annual prescriptions) of the total market. In comparison, Risperdal captured 14.2% of the world market in 1996, with a 69% increase in prescriptions. Haldol and haloperidol generics are still the most popularly prescribed anti-psychotics with 18.8% of the worldwide market and 3.1 million prescriptions (Pharmaceutical Approvals Monthly 1996).
Policy Interaction in Product Development and Distribution
The key legal issue surrounding Sandoz's marketing strategy was that the FDA required a patient management system, but they did not require that Sandoz should control the only patient management system thus creating a distribution monopoly and limiting the supply of the drug (Peck 1990). In August 1990, the FDA clarified their labeling requirements to explain that any patient monitoring system could be used with the drug as long as the system maintained certain standards to ensure patient safety (Peck 1990). Despite protests from provider groups such as the American Medical Association (AMA) and the American Pharmaceutical Association (APhA) who felt that pharmaceutical companies lacked the authority to credential pharmacy or medical practice, the FDA required Sandoz to be responsible for registering alternative patient monitoring systems (Martin 1991, Hospital and Community Psychiatry 1991).
In January, 1991, Sandoz separated the sale of clozapine from the monitoring system, which lowered the cost of the drug alone to $4160 per patient per year, provided that the supplier had their own patient monitoring system registered by Sandoz. Also at this time, Sandoz began a public relations advertising campaign in USA Today and the Wall Street Journal declaring that they had made no profit on Clozaril, and with the patent expiration date only 4 years away, were almost guaranteed not to make a profit. In May, 1991, HCFA(now known as CMS) ordered state Medicaid programs to cover the cost of clozapine. In June, 1991, the FTC pronounced that Sandoz had violated antitrust laws by requiring patients to enroll in an exclusive and extensive blood monitoring program. In 1992, Sandoz reached a $20 million settlement with the provider groups. Despite these lowered market barriers and the lower cost of clozapine, as of 1996 only 11,000 patients were receiving clozapine in the U.S..
Likely Future of the Product
Recently, the FDA Psychopharmacologic Drugs Advisory Committee unanimously recommended that after 6 months blood monitoring could be reduced to every two weeks and voted 7-3 to allow monitoring after 1 year of therapy to be voluntary. The FDA based their decision on a national registry study which showed that the risk of agranulocytosis is greatest in the first few months of therapy. This ruling will expand the market for clozapine treatment to include patients who did not use clozapine because of the monitoring requirements and will make treatment less expensive. Because the rate of agranulocytosis is low and can be identified and controlled, clozapine may yet become a popular first-line therapy (Pink Sheet 1997).
Clozapine faced a few other significant market barriers, all associated with the required patient monitoring that may be reduced now that the monitoring requirements have been lowered. These barriers included patient reluctance to use clozapine because of the required monitoring which was time consuming, involved needles, and required them to go to the doctor on a weekly basis (Boodman 1993). As the monitoring restrictions are reduced, more patients may be willing to try clozapine, potentially as a first-line therapy.
Clozapine's U.S. market exclusivity ended in 1995. Clozapine served as the first model of an antipsychotic agent without extrapyramidal side-effects, and thus spawned research into the newer classes of atypical anti-psychotics entering the market today. Since the introduction of clozapine, several atypical anti-psychotics have entered the market increasing competition. These products, e.g., risperidone, hope to demonstrate the same clinical efficacy as clozapine without the side-effects. Although clozapine has not reached its market potential of 250,000 patients, further clinical research based on second generation clozapine products that do not have a potentially fatal risk factor may be more successful in penetrating the market.
Conclusion
Clozapine had great clinical promise when first introduced in Europe in the 1960s. Once the potentially fatal side-effect of agranulocytosis was properly managed, the demand for clozapine rose but not to the level of initial expectations. The major market barrier to the success of clozapine in the U.S. was the prohibitively high cost of the associated patient monitoring system. This is the key issue of this case study that could be a potential market barrier for the development of cocaine pharmacotherapies in the future.
Case Study 4: Nicorette
Introduction
Drug Overview
Nicotine polacrilex is a smoking cessation therapy marketed under the trade name Nicorette by SmithKline Beecham in the United States and Pharmacia & Upjohn internationally for the treatment of nicotine addiction. The clinical effects of Nicorette allow the medication to serve as a temporary substitute for nicotine, slowly decreasing the patient's craving for nicotine. In contrast to the Nicoderm CQ transdermal patch, the Nicorette gum delivers a controlled dosage of nicotine to the body through the mucous membrane lining of the mouth (Nicorette 1997). Patients may obtain the medication over-the-counter at pharmacies or from their physicians. Clinical trials suggest that counseling and support received during the initial few weeks of treatment help patients cope with the behavioral aspects of nicotine addiction (Law 1995).
Market Overview
The potential size of the market for treatment of addiction to nicotine is significantly greater than that of other addictive substances such as heroin. According to a 1995 report by Abt Associates, approximately 496,000 people were addicted to opiates. In contrast, according to the Centers for Disease Control and Prevention, approximately forty-eight million Americans were current smokers in 1994. As Figure 32 highlights, the potential market for smoking cessation products has remained between forty-eight and fifty-five million people since 1965.
Figure 32: Total Number of Current Adult Smokers, 1965-1994
Key Issues from the Case Study
Similar to other pharmacotherapies for substance abuse addiction, government agencies have played an important role in the research and development of the medication. Government agencies financed many studies conducted in the late 1970s and early 1980s that led to FDA approval of Nicorette.
In contrast to most pharmacotherapies for substance abuse addiction, Nicorette is not subject to the different levels of federal, state, and local regulations and restrictions. The pharmaceutical companies involved in marketing Nicorette did not have to direct their time and energies toward rescheduling the medication or overcoming legislative barriers. Like more traditional medications, the medication was quickly introduced into the market after FDA approval.
A key difference between Nicorette and other medications for substance abuse addiction concerns the marketing strategies used by the pharmaceutical companies. SmithKline Beecham and Marion Merrell Dow (former licensee of Nicorette and now Hoechst Marion Roussel) were able to market the medication to the pharmacies and the physicians in the private sector. When Nicorette was approved as an over-the-counter smoking cessation therapy in 1996, the pharmaceutical companies were able to directly market the medication to smokers.
The following discussion provides a more comprehensive and detailed overview of the research, development, and marketing experiences of Nicorette.
Product History and Development Timeline
Figure 33: Timeline for Development, Approval, & Implementation of Nicorette
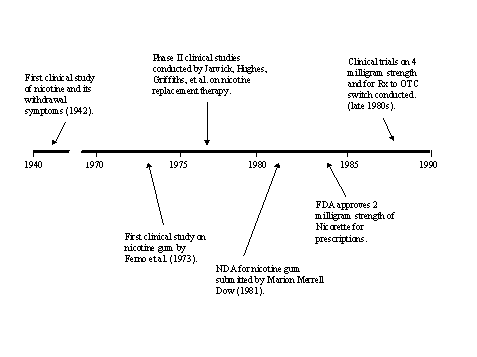
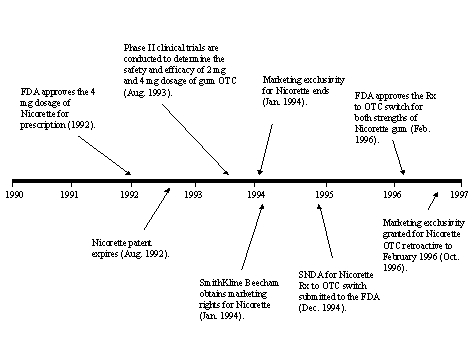
As the timeline in Figure 33 highlights, the first formal clinical study of nicotine and its withdrawal symptoms was conducted in 1942. However, the first clinical study on nicotine gum was not conducted until 1973 by Ferno et al. Clinical research on Nicorette and nicotine replacement therapy conducted throughout the 1970s and 1980s was financed jointly by the private and public sectors. In the early 1980s, Pharmacia & Upjohn were conducting clinical research that led to the development of nicotine polacrilex (i.e., Nicorette gum). The NDA for the 2 mg strength of Nicorette was submitted in 1983. In 1984, the medication was approved by the FDA as the first smoking cessation treatment therapy. In 1984, Marion Merrell Dow was licensed the rights to market the medication within the U.S. while Pharmacia & Upjohn marketed the medication abroad.
In 1992, the FDA approved the 4 mg strength of the medication and the patent on the compound expired. After obtaining FDA approval for the 4 mg medication, Marion Merrell Dow and SmithKline Beecham joined their "consumer product" lines. Marion Merrell Dow merged with SmithKline Beecham in an effort to halt the diminishing sales generated from Nicorette; the sales for the medications fell 17% between 1991 and 1992 due to the emergence of new nicotine replacement therapies. This merger served as the launching pad for the efforts to switch Nicorette from prescription medication to an over-the-counter (OTC) treatment. In February 1996, the FDA granted SmithKline Beecham the right to distribute the medication over the counter and granted a three-year market exclusivity on the OTC version until February 1999.
Clinical Development and Product Positioning
Clinical Trials, Phases I - III
As highlighted in the previous section, the first clinical trial that identified the withdrawal symptoms generated by the loss of nicotine was conducted in 1942 by Johnston et al. Intensive research on nicotine dependence and nicotine withdrawal symptoms, however, did not occur until the 1970s. During the early 1970s, researchers inferred nicotine dependence from titration studies, studies that confirmed the nicotine seeking behavior, and nicotine withdrawal symptoms (Henningfield 1997). This research, funded in part through grants from NIDA, the National Cancer Institute (NCI), and the National Heart, Lung and Blood Institute (NHLBI), laid the groundwork for researching nicotine substitution therapy and alternative delivery systems for nicotine. The initial nicotine replacement therapy was investigated in the early 1970s by Ferno et al. (Nunn-Thompson 1989). Based upon the hypothesis that nicotine was the agent that caused the smoking dependence, Ferno sought to develop a mechanism for nicotine replacement to help reduce the characteristic craving for cigarettes. In 1973, Ferno et al. developed the chewing gum formulation (nicotine polacrilex) that would give the body reduced amounts of nicotine until the dependence on nicotine was eliminated. Research on this replacement therapy continued throughout the 1970s and received top priority from the FDA.
NDA Applications
In 1981, Pharmacia & Upjohn submitted the NDA for nicotine polacrilex; the NDA for the 2 mg strength gum was subsequently approved in 1983 (Nunn-Thompson 1989). After obtaining FDA approval, Pharmacia & Upjohn licensed the rights of Nicorette to Marion Merrell Dow to market the medication in the United States. Research on nicotine replacement systems continued by NIDA supported researchers including Jarvick, Hughes, Griffiths, and Henningfield (Henningfield 1997). Throughout the 1980s, research on Nicorette gum continued. Clinical trials were conducted throughout the 1980s that determined that nicotine replacement therapy was more effective when accompanied by behavioral adjuncts including counseling, nicotine weaning, and relapse prevention. According to clinical trials conducted through 1985, patients that received nicotine polacrilex gum had 27% abstinence rates with intensive specialized support, 21% with minimal support, and 10% when the medication was taken at the advice of a physician.
Concurrent to the research on the efficacy of nicotine gum in clinical settings, Marion Merrell Dow and Pharmacia & Upjohn were conducting research on the efficacy of 4 mg doses versus 2 mg doses of Nicorette. In 1992, the FDA approved the 4 mg dose of Nicorette gum in conjunction with the approval of four other nicotine replacement therapies including Nicoderm CQ and Nicotrol nicotine transdermal patches.
SmithKline Beecham Merger and the OTC Research
The merger between SmithKline Beecham and Marion Merrell Dow consumer lines in 1992 initiated the clinical research for the potential switch of Nicorette from prescription medication to an OTC remedy (The Tan Sheet 1993). Clinical trials on the efficacy and safety of the OTC product were conducted throughout the spring of 1993 and were completed in August 1993. Research continued in 1994 when SmithKline Beecham (which had obtained marketing rights to Nicorette as of January 1, 1994) submitted the SNDA. In the SNDA, SmithKline included a non-clinical study on the comprehensiveness of the labeling to consumers, two clinical studies that examined physician's prescription rate of the medication and the patients' subsequent quit rates, and two OTC use trials. FDA determined that these clinical trials demonstrated the safety and efficacy of the OTC product and FDA approved the Rx-to-OTC switch in February 1996. In October 1996, the FDA granted the product 3 years of market exclusivity under the Waxman-Hatch regulations retroactive to the OTC approval date (The Tan Sheet 1996). The Waxman-Hatch regulations were established under the Drug Price Competition and Patent Restoration Act of 1984 and these regulations set the criteria for drugs (prescription and OTC) seeking market exclusivity. To obtain market exclusivity, the following criteria must be met:
- The FDA must not have approved an identical drug product with the same conditions of use prior to this request.
- The NDA or SNDA must contain reports of new clinical investigations, not studies relied on for the approval of another drug product or a previous NDA.
- NDAs must be supported by clinical studies other than bioavailability studies.
- The clinical studies in the NDA must be conducted or sponsored by the person submitting the application and the studies must be essential to the approval of the NDA.
Product Marketing Strategy and Sales
In contrast to other pharmacotherapies to substance abuse addiction, the manufacturers of Nicorette did not have additional significant market barriers after FDA approval. After the approval of the prescription strength form of Nicorette, Marion Merrell Dow focused its energies on the physicians and the pharmacies. The company educated physicians and pharmacists concerning the efficacy of the medication when taken in conjunction with physician support and behavioral counseling.
Marketing Strategy
Following the OTC approval of Nicorette gum, SmithKline launched a new marketing strategy directed at consumers. SmithKline utilized numerous different approaches to educate the public about the new medication, including the following:
- Commercial advertisements in the media. In April 1996, SmithKline Beecham launched television advertisements that featured portraits of quitters that had relied upon Nicorette to stop smoking. The advertisements were 30-second spots that aired during prime-time programming.
- Nicorette Webpage. SmithKline Beecham created a webpage about its medication for smokers that surfed the web. The webpage educated potential consumers about the medication, provided "helpful hints" for smokers, and had a dependency quiz that potential consumers should take prior to purchasing Nicorette. In addition, the webpage linked to the company's Committed Quitters Program, "a personalized 12-week smoking cessation program that will . . .keep you on the road to quitting."
- Creation of a partnership with the American Lung Association. SmithKline gave the American Lung Association a grant to establish a hotline entitled "Counselors on Call." This hotline operated from April 17 - 26, 1996, to coincide with the launch of the OTC version of Nicorette. Smokers that contacted the hotline received counseling from ALA officials who provided information on smoking cessation including the use of Nicorette (The Tan Sheet 1996).
Marketing Sales
As highlighted by the table below, the sales of Nicorette increased dramatically after the FDA approval of the OTC version of the medication. Sales in the second quarter of 1996 increased over 100% from the sales in the first quarter, rising from $37 million to $78 million. During 1996 alone, the sales from Nicorette surpassed $225 million dollars. This figure is comparable to target peak annual revenues for new drugs of major pharmaceutical companies. Nicorette is sold at an average wholesale price of $3.77 per day (2 mg strength) and $6.59 per day (4 mg strength), and retails in pharmacies in the range of $45 to $55 for a 108-count package of Nicorette and $25 to $30 for a 48-unit package (Garrett 1994; Tan Sheet 1996). By comparison, the average wholesale price of $3.77 per day for Nicorette (2 mg strength) is more expensive than methadone ($0.50 per day) and LAAM ($2.00 per day), but is less expensive than naltrexone ($4.50 per day).
In terms of market share, Nicorette currently dominates the smoking cessation treatment product market; Nicorette accounts for 81 percent of the market, while its closest competitor, Nicoderm CQ only accounts for 6.5 percent of the market (Tan Sheet 1997).
Figure 34: Nicorette Sales in 1996 in U.S. Market
Policy Interaction in Product Development and Distribution
As discussed above, the federal government played an important role in the development of the medication. Grants from NCI, NIDA, and NLBHI funded, in part, research that laid the groundwork for the development of nicotine replacement therapies. After Ferno et al. developed the nicotine polacrilex gum, the FDA granted this research top priority (Nunn-Thompson 1987).
During the early 1980s, the FDA acknowledged the emergence of treatment systems for smoking cessation. In response to the research on these treatments, the FDA established conditions under which OTC smoking deterrent drug products would be recognized as safe and effective. In its proposed conditions, the FDA acknowledged that a single protocol for the evaluation of smoking deterrent products is not necessarily universally appropriate. However, the FDA also acknowledged that "it is imperative that well-controlled clinical trials be performed to evaluate these drugs. In designing these trials, important issues must be considered carefully in order to ensure proper evaluation" (Federal Register 1982). The guidelines and the conditions established by this panel of officials shaped the clinical trials that were eventually conducted on Nicorette and other smoking cessation products approved for OTC use.
Likely Future of the Product
Currently, the 2 mg and 4 mg strengths of Nicorette are available to consumers as OTC products. In February 1996, Nicorette obtained market exclusivity for their product under the Waxman-Hatch regulations; this market exclusivity will expire as of February 1999. Phase IV clinical trials are currently being conducted by SmithKline Beecham on safety of the medication in adolescents; it is uncertain whether these clinical trials will be employed to get a new indication for the medication.
Currently, three smoking cessation products, Nicorette gum (SmithKline Beecham), Nicoderm CQ patch (SmithKline), and Nicotrol patch (McNeil), have been approved by the FDA for OTC use. Product development continues and new methods of delivery (i.e. inhalers) have recently been approved by the FDA for prescription use. Once the market exclusivity on Nicorette, granted as of February 1996, expires in February 1999, SmithKline is likely to see other comparable products vying for market share.
Conclusion
As discussed above, the market for smoking cessation products differs significantly from the markets for medications such as LAAM or naltrexone. First, the size of the potential market varies tremendously. According to the Centers for Disease Control and Prevention (CDC), nearly 48 million people identified themselves as current smokers in 1994. In 1995, there were only 496,000 individuals that were identified as heroin addicts. The potential return on investment is higher in the market for smoking cessation in part due to its tremendous size. Second, manufacturers of Nicorette did not have to overcome regulatory barriers (i.e. rescheduling of the medication) after FDA approval, and did not have to adhere to DEA regulations. Once Nicorette was approved, the drug companies were able to initiate their marketing strategies throughout the country. Third, SmithKline Beecham and Marion Merrell Dow were able to directly market the medication to physicians in the private and public sectors and to pharmacies; their marketing strategies were not limited to a select number of highly regulated clinics.
These differences have significantly influenced the level of market penetration obtained by the smoking cessation products versus the market penetration of other pharmacotherapies for substance abuse addiction. In the past 15 years since its initial approval, Nicorette has been taken by over 13 million people in the United States alone (The Tan Sheet 1996).
Market Barriers to the Development of COCAINE Pharmacotherapies
The purpose of this section is to characterize private industry's views on market barriers to anti-addiction pharmacotherapy development. The 1995 IOM report, Development of Medications for the Treatment of Opiate and Cocaine Addictions, identified numerous market obstacles that appear to hamper private sector investment in anti-addiction medication development, e.g., market size, development costs, regulatory requirements for approval, and social attitudes about drug abuse (see Figure 35). This section pulls together industry views on these and other market barriers to pharmacotherapies for cocaine abuse and addiction with the objective of identifying barriers with the greatest impact on industry investment decisions. Additional information gathered for this section provides insights into estimating the relative import of various market barriers, including those considered to be "make-or-break" or otherwise critical barriers, and of the feasibility of surmounting them under current circumstances.
Figure 35: Market Obstacles & Creating Incentive
Source: IOM, Development of Medications for the Treatment of Opiate and Cocaine Addictions, 1995.
This section contains information from four sources: a) interviews with executives from five private firms (three pharmaceutical and two venture capital firms); b) the market analysis for a prospective cocaine medication; and c) scenarios of company decisionmaking; and d) case study reports of LAAM, naltrexone, clozapine, and Nicorette. The case study reports, market analysis, and scenarios of company decisionmaking provide information about the market potential for cocaine medications and lessons on development and marketing from previously developed pharmacotherapies.
The interviews with executives of private firms helped to identify the barriers to industry development of medications for cocaine abuse and addiction. It is important to note that the small sample of firms precludes either a representative study or assurance that all possible barriers have been identified. Instead, the primary goal of the interviews with the executives of private firms was to better understand the relative significance of barriers to this market.
Industry Evaluation of New Drug Investment Opportunities
In order to provide context for the market environment and background for understanding the market barriers from industry's perspective, this section summarizes pharmaceutical and venture capital firms' approaches to the evaluation of new drug investments.
Pharmaceutical Evaluation of New Drug Investment Opportunities
As discussed above, a pharmaceutical company's decision to develop and market a drug is predicated on a number of factors related to the probability of economic success of a given product. Each of the several companies that were interviewed for this study indicated that the evaluation of a candidate drug project hinged on an overall assessment of risk versus reward. Indeed, several of our interviewees reported using some form of risk-reward analysis as the primary analytic framework to inform drug development decisions.
The decision to develop a drug requires the input of both the clinical development departments of the company and the market research/product planning departments to help assess the clinical and commercial viability of candidate products. One of the company executives described a two-step decision making process. First, a pre-clinical committee meets to discuss key topic areas of interest for the company (e.g., CNS, cardiovascular disease) and then agrees upon a preferred list of development topics. For example, if there is a research breakthrough in the area of cocaine abuse and addiction, the pre-clinical committee first would have to agree that the breakthrough warranted the company's focus on that particular disease area. If the pre-clinical committee agrees that drug development should be pursued, it would refer the drug candidate to the commercial development arm of the company, which then would assess the product's market potential.
A part of the overall risk-versus-reward analysis by pharmaceutical firms involves financial calculations, e.g., the net present value (NPV) of a product. Although these calculations may be performed at any point during the development cycle of the drug, one interview respondent noted that most financial calculations are performed at the end of Phase II or the beginning of Phase III clinical trials of a product. These financial calculations are most often performed by market research or product planning departments as part of an assessment of a candidate drug's potential commercial viability. Interview respondents cautioned that while NPV and other financial calculations are important quantitative indices of risk and reward, certain qualitative factors that may be more difficult to quantify (e.g., product liability issues, opportunities to pursue drug development in other areas) must be weighed in assessing risk versus reward.
Venture Capital Evaluation of New Drug Investment Opportunities
Venture capital firms (VCs) reported market potential (e.g., projected revenues) and probability of financial success as the main factors guiding investment decisions on new products. VC interviewees suggested that the evaluation of pharmacotherapies is no different from that of any other product or business plan. These companies are most interested in high financial return, and typically are less concerned about image or altruistic interests of shareholders.
The VC interviewees were not involved primarily in pharmacotherapies for substance abuse. They attributed this not to a lack of interest, but rather to a lack of opportunities. One VC company executive indicated that in his five years of evaluating biotechnology and pharmaceutical products, he had never seen or evaluated a compound for the treatment of addiction.
VCs reported that their evaluation of candidate investments did not focus on a "magic number" such as a return on investment (ROI) calculation. Rather, VCs employ a large number of criteria to screen potential investment opportunities. One large venture capital firm's investment standards (noted in the recent literature, not drawn from interviews) are based on four fundamental questions about the candidate investment:
- How big?- refers to the size of the market multiplied by the market penetration that the proposed company is likely to achieve. Market penetration factors in competition, market trends, and potential for a family of related products to result from the product, e.g., a diagnostic kit for a disease and a treatment for the same disease.
- How fast?- refers to the time necessary to develop the product, obtain regulatory approval, and market a product.
- How much?- refers to estimates of the total capital required before a company achieves cash-flow break-even point, and to determination of the source of capital, e.g., VCs, banks, government grants, corporations.
- How can do?- refers to overall assessment of the investment opportunity. Includes factors such as projected revenues, time and capital needed to break even, differentiation of product from competitor products, and quality of people involved (e.g., skills, expertise, reputation in field)
(Adapted from: Kunze B., Nothing Ventured, 1993).
One VC interviewee noted criteria similar to those noted above when evaluating a pharmaceutical or biotechnology investment. One of the VC firms analyzes potential ventures by considering all facets of market potential, including pharmacology, data on how the molecule works, animal models, results from clinical trials (if available), and potential revenues. In the case of a pharmacotherapy for cocaine abuse and addiction, this VC firm is most concerned about the following: (1) the likelihood of a "clean" trial (e.g., a trial whose results are not confounded by lack of patient compliance, co-morbidities, dropouts, and other related factors) may be low, (2) compliance issues of the drug post-market, and (3) lack of clarity of the desirable clinical treatment endpoints, confounding judgments about whether an anti-addiction drug works. However, the VC representative stated that social stigma and other qualitative factors would not be an issue if the projected revenues for the drug appeared to be strong.
Another VC interviewee stated that the firm used similar investment criteria, adding to the key drug investment criteria the importance of the reputation and quality of the people involved in the venture, and ease of regulatory approval.
Industry Perception of Market Barriers
Some of the market barriers identified in the 1995 IOM report were confirmed through the interviews with private firms and case studies conducted for this effort. Figure 36 summarizes the market barriers that were confirmed (in full or in part) and not confirmed during this study. (As noted above, the number of interviews and case studies was limited by the scope of this study.) Although no new general types of market barriers were identified during this project, certain ones were elaborated or described in a more contemporary context.
| Market Barrier from IOM Report | Confirmed | Not Confirmed | Notes |
|---|---|---|---|
| Uncertain market environment | ✓ | ||
| Discovery | |||
| Limited number of researchers focusing on drug abuse | ✓ | ||
| Lack of well-characterized animal models of cocaine addiction | ✓ | ||
| Limited basic science knowledge of addiction, craving, and relapse | ✓ | ||
| File IND | |||
| Clinical Studies | |||
| DEA regulations | ✓ | ||
| Complications of concomitant illness and polydrug abuse | ✓ | ||
| Patient population perceived as difficult to study | ✓ | ||
| Efficacy outcomes difficult to define or measure | ✓ | ||
| Few clinical investigators | ✓ | ||
| File NDA | |||
| Length of FDA approval process | ✓ | ||
| Other Approval | |||
| State rescheduling | ✓ | ||
| Varied state / local approval processes | ✓ | ||
| DEA review time | ✓ | ||
| Marketing | |||
| Varied state and local regulations | ✓ | ||
| Lack of traditional marketing to physicians | ✓ | ||
| Pricing clause in DHHS CRADAs | ✓ | NIH recently removed reasonable pricing clause from CRADAs | |
| Small foreign market | ✓ | ||
| Treatment System | |||
| Limited number of narcotic treatment programs | ✓ | ||
| Stigma of drug-abuse | ✓ | ||
| Bias by some treatment providers against pharmacologic treatments | ✓ | ||
| Varied state / local treatment regulations and financing mechanisms | ✓ | ||
| Uncertain treatment financing | ✓ | ||
| Adapted from: IOM, Development of Medications for the Treatment of Opiate and Cocaine Addictions, 1995. | |||
Two types of market barriers emerged from our report. Critical barriers are those that must be surmounted in order for pharmaceutical firms to regard as feasible the prospects for developing cocaine addiction medications that will be financially successful. Non-critical market barriers are those that, if lowered or eliminated, may enhance (though perhaps only marginally) the financial outlook for developing cocaine addiction medications only if the critical barriers are also lowered. That is, without lowering the critical barriers, lowering the non-critical ones would be unlikely to transform an unattractive market into an attractive one.
Among the diverse market barriers perceived by the industry, three emerged as critical in this study, i.e., that would have to be lowered or eliminated in order to begin to make new drug development attractive to pharmaceutical companies:
- Small and uncertain market for cocaine addiction and abuse pharmacotherapy
- A substance abuse treatment system that limits access to this market
- Limited and uncertain payment for treatment
The beginning of the following section briefly describes private industry's views on the strength and importance of these three critical market barriers. The remainder of the section describes non-critical market barriers relative to the process of drug development and marketing.
Critical Barrier 1: Small and Uncertain Market for Cocaine Addiction and Abuse Pharmacotherapy
The small size and uncertainty of the market for cocaine pharmacotherapies constitutes a critical barrier to development of a cocaine abuse pharmacotherapy. Although all of our interviewees agreed that the total number of cocaine users is large, they recognized that the market for a cocaine abuse treatment is likely to be much smaller than the absolute number of people that use cocaine. Representatives of one pharmaceutical company said that they used a conservative estimate of the market size for cocaine treatment that was about half of the generally accepted 2 million heavy users.
Uncertain market penetration was another reason for the skepticism of the market. Interviewees from two of the companies stressed that potential patient compliance problems and limited access to patients (i.e., given the need for many substance abusers to obtain pharmacotherapy under controlled settings) made them uncertain about the true market size for cocaine treatment. Representatives of two pharmaceutical companies noted that many publicly-funded treatment centers were managed by non-physicians who tended to oppose the use of drugs to treat substance abuse, which such staff regarded as "behavioral" conditions, thereby further restricting the potential sale of these drugs.
Critical Barrier 2: A Substance Abuse Treatment System that Limits Access to this Market
There was consensus among the pharmaceutical company representatives that the current substance abuse treatment system constitutes a great market barrier that severely limits opportunities for market penetration.
As reported in the case studies, sales of LAAM and naltrexone were restricted by the limited number of heroin and alcohol treatment programs and the limited capacity of these programs. Twenty-five percent of opiate addicts receive treatment from the methadone maintenance programs while less than 1 percent of people in the U.S. afflicted by alcohol abuse and dependence are in alcohol treatment centers. LAAM is restricted to maintenance programs as required by The Narcotic Addict Treatment Act of 1974. Distribution of naltrexone is limited to comprehensive treatment centers, which enhance patient compliance. These market restrictions severely limit sales of the drugs and create another barrier to pharmaceutical companies that are considering developing a pharmacotherapy for drug addiction. One major market advantage of Nicorette is that, with an over-the-counter formulation, patients do not need to visit a treatment center or a provider to obtain treatment, vastly expanding market potential.
The lack of medical treatment models is another shortcoming of the substance abuse treatment system that poses a major concern for pharmaceutical companies. The pharmaceutical company executives cited an "anti-medication" climate among publicly-funded treatment center staff that would severely limit sales of pharmacotherapies through treatment centers. Interviewees implicated the large number of non-physicians (a.k.a. "non-prescribers") as the main reason for the anti-medication sentiment at these treatment centers.
Our market analysis supports the finding that there is a large number of non-physicians at publicly-funded treatment centers. The 1991 NDATUS study of specialty substance abuse providers surveyed 9,000 treatment centers and found that there were only about 2.2 full-time equivalent psychiatrists and other physicians, respectively, per 1,000 enrolled patients (Office of Applied Studies, 1993). The most recent surveys that have examined staffing patterns confirm that the substance abuse treatment system involves little or no physician time in the treatment of patients.
For example, methadone treatment for heroin addiction would appear to be the most medically oriented model of drug treatment. However, the role of physicians in methadone clinics is generally small and circumscribed to initial diagnostic assessments (i.e., of heroin addiction), management of methadone dosage, and some primary health care services. Most clinic services are oriented to the behavioral and psychosocial needs of the patients, and are delivered by counselors, social workers, and, less often, psychologists (Institute of Medicine, 1990).
This sentiment was repeated in our LAAM and naltrexone (Trexan) case studies, which found that a major market barrier for both products has been that treatment decisions and funding for heroin addiction are often controlled by state-level substance abuse program administrators who often do not have clinical backgrounds.
Critical Barrier 3: Limited and Uncertain Payment for Treatment
Another critical market barrier is the uncertainty surrounding the reimbursement of cocaine abuse and addiction treatment. The pharmaceutical company respondents, as well as one from a VC firm, voiced their concern over the heavy reliance of the substance abuse market on federal and state government reimbursement. The perception among the drug companies is that many cocaine addicts do not have private insurance and rely on Medicaid for treatment, and that only a portion of those individuals with private insurance use their benefits for drug abuse treatment. This perception is consistent with the 1995 TEDS data (described earlier in report) that found over 68 percent of enrolled cocaine abusers had no health insurance, and an additional 17 percent had Medicaid coverage. One pharmaceutical company noted that substance abuse services continue to be subsumed under mental health benefits of entitlement programs, and that the overall budget for mental health services continues to shrink in light of other competing health priorities.
Reimbursement was an issue for LAAM, naltrexone, and clozapine. Treatment for heroin addiction (e.g., LAAM and naltrexone) has been funded primarily through federal and state budgets, making reimbursement difficult for pharmaceutical companies. In the case of clozapine, many public payers (e.g., Veteran's Administration, several state Medicaid agencies) refused to pay the additional cost of purchasing the Clozaril Patient Monitoring System ($9,000) before Sandoz uncoupled the drug and the monitoring system.
As reported in our market analysis, price sensitivity to a cocaine medication is another aspect of payment that may be a critical market barrier because price resistance may limit market size. As no approved pharmacotherapy for cocaine abuse has been tested on the market, it is not possible to know how sensitive the market would be to such a medication. However, indirect available evidence from other substance abuse medications (e.g., LAAM, naltrexone) and the current nature of cocaine abuse treatment and its financing would appear to indicate that the market would be very sensitive to the price of a cocaine medication. In a market where the average daily treatment cost is a modest $9.00 per outpatient and $23.00 per inpatient, a cocaine pharmacotherapy priced at a daily dose of a few dollars would represent a significant proportionate cost increase. This may be particularly so in the estimation of substance abuse treatment providers that are vested in psychosocial approaches to the exclusion of pharmacotherapy. It is important to note that the price sensitivity of the current treatment system may vary considerably from that of more typical pharmacotherapy markets that involve physician prescribing and distribution through pharmacies.
Non-Critical Market Barriers
Regulatory Issues for Approval: File IND/Clinical Studies, File NDA, and Other Approval
Several points within the drug approval process were seen by industry as potential market barriers to product development in this field. However, as a group, none were viewed as major barriers that could not be surmounted, especially if the market barriers cited as critical ones above were lowered or eliminated.
For two of the pharmaceutical companies interviewed, the cost of funding the necessary clinical trials for obtaining FDA approval was seen as a minor barrier to cocaine pharmacotherapy development. However, the pharmaceutical company that reported that the science base was not a barrier also reported that the regulatory aspects of development would not preclude moving ahead, so long as the science base and the financial market potential were evident. Also, one of the VC firms interviewed stated that drug regulation would not be a barrier if the market potential of the drug was very favorable.
Patient Populations Perceived to Be Difficult to Study
Three of the four case study drugs were for patient populations perceived to be difficult to study for a variety of reasons (e.g., patient recruitment, compliance, and co-morbidities). For example, patient compliance has been seen as a barrier to the success of naltrexone in both the heroin addiction and alcoholism markets, because the drug is not effective unless patients take part in a treatment program with a more intensive psychosocial component than for other pharmacotherapies. Compliance is often an issue when treating patients with schizophrenia, primarily because they may not recognize their illnesses or understand the need for treatment. In addition, alcoholics and other substance abusers may also have severe co-morbidities (e.g., hepatitis or depression), which may lead to poor clinical trial outcomes or adverse events that are unrelated, but wrongly attributed, to the study medication. Finally, researchers involved in the development of LAAM and naltrexone had difficulty recruiting patients because methadone maintenance clinics were unwilling to refer patients to clinical trials for fear of lowering their patient census and associated reimbursement.
Representatives of two companies stressed that cocaine abuse and addiction drugs in the pipeline need better access to patients for conducting clinical trials. A representative of another company described how adverse effects experienced by cocaine patients with multiple co-morbidities could be improperly attributed to its investigative treatments. Although pharmaceutical company representatives viewed these patient-related difficulties as real problems, they did not regard these problems as absolute barriers that could not be overcome given other incentives for entering the market.
Ambiguity of Desired Clinical Endpoints
The uncertainty associated with designation of required clinical endpoints to be used in clinical trials of medications for cocaine addiction was cited as a market barrier, though not a major one. Two pharmaceutical company interviewees identified this ambiguity as a potential barrier, and one company representative expressed some concern that "chasing a moving target" could increase the costs of conducting clinical trials. However, the pharmaceutical company interviewees were not aware of the FDA's current efforts to update its draft guidance for trials of drugs to treat cocaine addiction.
The case study of naltrexone demonstrated the difficulty of convincing providers and patients that a reduction in use of heroin or alcohol can result in favorable health outcomes. Although naltrexone blocks the effects of both heroin and alcohol, it does not prevent patients from using these substances. Researchers noticed that because patients using naltrexone did not experience the euphoric effects of heroin or alcohol, they had less incentive to inject heroin or drink alcohol, and their volume of use was reduced. Many provider and patient support groups have expressed that total abstinence is the only acceptable cure.
DEA Regulation
DEA regulation was not generally cited by pharmaceutical company representatives as a market barrier. One interviewee mentioned the potential risk of exposing an existing successful product used for other indications to cocaine treatment. The company indicated that the increase in development costs of the drug and the potential for rescheduling of the drug could restrict the market opportunities for its original indication and market.
The case study of LAAM, which is regulated by the DEA as a Schedule II drug, demonstrates how DEA regulation can be a hurdle for drug development. This regulation places severe restrictions on distribution channels, primarily in order to prevent the drug from being diverted to the black market. DEA scheduling prevents a drug from being marketed in a state until that state has rescheduled the drug, and state rescheduling could delay product marketing by years.
It is highly unlikely that an existing product would meet the criteria for being rescheduled by the DEA. In order for a currently marketed product to be rescheduled because of its use in cocaine dependent persons, it would have to meet two criteria: 1) the drug must have abuse liability, and 2) the drug must be classified as a narcotic. While methadone and LAAM meet these criteria, naltrexone is not scheduled. In addition, products with abuse potential are scheduled at the time of their NDA approval, and if an existing product showed higher or lower than expected abuse liability after marketing, it could be rescheduled upwards or downwards, regardless of whether it ever was used in a cocaine dependent population (Cummings, 1997).
Other Approval Issues
The clozapine case study demonstrates that special requirements for approved use and other atypical restrictions can pose significant market barriers. Clozapine has a potentially fatal side-effect, agranulocytosis, that warrants strict patient monitoring. This contributed to clozapine's being approved as a second- or third-line therapy, i.e., for patients who are resistant to other treatments. The strict weekly monitoring of patients on clozapine increases the cost of treatment. Sandoz originally linked sales of the drug to its Clozaril Patient Monitoring System at a cost of almost $9,000 per patient per year, severely impeding the ability and willingness of payers to purchase this treatment and dampening sales.
Drug Marketing Issues
Two market barriers related to drug marketing were identified. As previously described, the possibility of the pharmacotherapy being distributed through publicly-funded treatment centers rather than through physician offices was a concern of the pharmaceutical companies because of the companies' limited access to patients.
Variations in federal, state and local regulations have proven to be market barriers in the case of LAAM. LAAM is the most highly regulated of the drugs (DEA Schedule 2), whereas Nicorette is the least regulated (available over-the-counter). DEA regulation of LAAM has limited market penetration by restricting delivery of LAAM to methadone maintenance clinics. In contrast, with its far less stringent regulation, Nicorette is readily available to its large target population.
Social Stigma
Social stigma of drug abusers was cited as a market barrier by representatives of two of the pharmaceutical companies, though it is regarded as surmountable if the commercial and scientific viability of the product is favorable. The pharmaceutical companies that identified social stigma as a barrier were familiar and sympathetic to the case experience of Eli Lilly and methadone, in which the drug's original analgesia market plummeted after people associated methadone with heroin addiction treatment.
Representatives of one of the pharmaceutical companies that has a strong CNS portfolio expressed concern that the negative image of the drug abuse population would hinder the likelihood of a pharmacotherapy's financial success. Further, one company representative indicated that if a highly promising or approved drug for a non-substance abuse CNS disorder showed potential effectiveness for treating cocaine addiction, the company would be very circumspect about pursuing the cocaine indication for fear that any adverse events or stigma associated with the substance abuse indication would threaten the market for the original indication.
One pharmaceutical company interviewee suggested that social stigma can be circumvented by renaming drugs for cocaine indications. Another interviewee suggested that future progress of the science base and pharmacology may enable designating different drugs from a class of closely related yet distinct molecules, all of which would have same or similar CNS actions. Thus, one molecule could be designated as a cocaine medication while another from the same class could be designated for another CNS indication, thereby avoiding the problem of attempting to market a molecule for a cocaine indication that is effective for another CNS indication.
Industry Perception of Science Base Readiness
There was a divergence of opinion among the pharmaceutical company interviewees about the readiness of the science base for cocaine pharmacotherapies. Representatives of two companies expressed skepticism about the readiness of the science base. One representative indicated that current limitations stem from a lack of understanding regarding the biological and genetic basis of addiction. The interviewee contended that, in contrast to the situation for Parkinson's disease, researchers have been unable to implicate the genetic abnormalities underlying addiction, and that the science base for cocaine abuse and addiction is "not close." This interviewee regarded the existing cocaine pharmacotherapies as "half-way technologies." Furthermore, scientists from the same company judged that the probability of a scientific breakthrough in the area of cocaine abuse and addiction in the near future is very low.
Representatives of another pharmaceutical company indicated that a financially successful cocaine medication needed to demonstrate long-term efficacy, but reported that the current science base for achieving long-term efficacy is "very weak." In contrast, representatives of another pharmaceutical company indicated strongly that the science base is ready, and consequently that the science base is no longer a market barrier to development of cocaine pharmacotherapies. This company reported that it had successfully identified several drug candidates that exhibited cocaine blocking activities in both in vivo and in vitro models.
Scientific executives from one company who questioned the science base did suggest that there are opportunities for existing and potential products to be used as effective adjuncts to cocaine abuse therapy. For example, existing drugs for anxiety could help manage symptoms associated with withdrawal.
The views cited in this study concerning the readiness of the science base come from personnel who are knowledgeable about drug development and marketing and are in decision-making roles in companies with real or potential interest in this field. However, these views are taken from a limited sample of such personnel.
Conclusions
Principal Conclusions
Under current conditions, pursuing development of a new cocaine pharmacotherapy via a typical full product development cycle is not economically viable from the standpoint of industry. Although a variety of hurdles or procedural impediments may affect prospects for new pharmacotherapy development in this area, most of these are regarded as surmountable by the industry. However, three critical market barriers to significant progress in bringing an effective pharmacotherapy to a viable market are:
- Small and uncertain market for cocaine addiction and abuse pharmacotherapy
- A substance abuse treatment system that limits access to this market
- Limited and uncertain payment for pharmacotherapy for this indication
Critical Market Barriers
Many of the market barriers identified in the 1995 IOM report were confirmed through the sources used for this study. Although no new general types of new market barriers were identified in this study, certain ones were elaborated or described in a more contemporary context.
Two main categories of market barriers emerged from this study. Critical barriers are those that must be lowered or eliminated in order for pharmaceutical firms to regard the prospects for developing cocaine addiction medications as financially feasible. Non-critical market barriers are those that, if lowered or eliminated, may enhance, though perhaps only marginally, the financial outlook for developing cocaine addiction medications only if the critical barriers are also lowered. That is, without movement on the critical barriers, lowering non-critical ones would be unlikely to transform an otherwise unattractive market into an attractive one.
Among the diverse market barriers perceived by the industry, three emerged as critical in this study, i.e., those that would have to be lowered or eliminated in order to begin to make new drug development attractive to pharmaceutical companies:
- Small and uncertain market for cocaine addiction and abuse pharmacotherapy
- A substance abuse treatment system that limits access to this market
- Limited and uncertain payment for pharmacotherapy for this indication
Critical Barrier 1: Small and Uncertain Market for Cocaine Addiction and Abuse Pharmacotherapy
The small size and uncertainty of the market for cocaine pharmacotherapies constitutes a critical barrier to development of a cocaine abuse pharmacotherapy. Although all of the company executives interviewed for this study agreed that the total number of cocaine users is appreciable, they recognized that the feasible market for a cocaine abuse treatment is likely to be much smaller than the absolute number of people that use cocaine. Representatives of one pharmaceutical company use a conservative estimate of the number of heavy cocaine users that is about half of the level of 2 million cited in this report.
Uncertain market penetration was another reason for the skepticism in industry. Interviewees stressed that potential patient compliance problems and limited access to patients made them uncertain about the true market size for cocaine treatment. Representatives of two companies noted that most publicly-funded treatment centers are managed by non-physicians who tend to oppose the use of drugs to treat substance abuse, which such staff regard as a "behavioral" condition, thereby further restricting the potential sale of these drugs.
Critical Barrier 2: A Substance Abuse Treatment System that Limits Access to the Market
There are multiple, interrelated aspects of the current substance abuse treatment system that limit the market prospects for any new pharmacotherapy for cocaine addiction. These limitations are apparent in the case studies, were raised by company executives interviewed for this study, and are corroborated by modeling of certain scenarios. Sales of LAAM and naltrexone were restricted by the limited number of heroin and alcohol treatment programs and the limited capacity of these programs. Whereas 25 percent of opiate addicts receive treatment from the methadone maintenance programs, only about 5 percent of those afflicted by alcohol abuse and dependence are in alcohol treatment centers. Distribution of LAAM is restricted to maintenance programs as required by The Narcotic Addict Treatment Act of 1974. Prescription of naltrexone is recommended to be linked to enrollment in comprehensive treatment centers in order to improve patient outcomes. In contrast, because Nicorette is an over-the-counter formulation, patients need not visit a treatment center or a provider to obtain treatment, vastly expanding the drug's potential market.
The lack of medical treatment models in substance abuse treatment centers contributes to their being a critical market barrier. Pharmaceutical company executives cited an "anti-medication" climate among the publicly-funded treatment center staff that would severely limit sales of pharmacotherapies through treatment centers. Interviewees indicated that the large number of non-physicians (sometimes referred to as "non-prescribers") at treatment centers often have strong anti-medication sentiments. As noted above, recent surveys that have examined staffing patterns confirm that the substance abuse treatment system involves little or no physician time in the treatment of patients. This observation was confirmed in the LAAM and naltrexone (Trexan) case studies, which found that treatment decisions and funding for heroin addiction are often mediated by state-level substance abuse program administrators who often do not have clinical backgrounds.
Critical Barrier 3: Limited and Uncertain Payment for Pharmacotherapy
Industry decision makers recognize the heavy reliance of the substance abuse market on federal, state, and local government reimbursement. The perception among the drug companies is that many cocaine addicts do not have private insurance and rely on federal and state government sources for treatment, and that only a portion of those individuals with private insurance use their benefits for drug abuse treatment. One executive noted that substance abuse services continue to be subsumed under mental health benefits of entitlement programs, and that the overall budget for mental health services continues to shrink in light of other competing health priorities.
Payment status is a recognized barrier for LAAM, naltrexone, and clozapine. Treatment for heroin addiction (e.g., LAAM and naltrexone) has been funded primarily through federal and state budgets, making reimbursement difficult for pharmaceutical companies. As noted above, price sensitivity to a cocaine medication is another aspect of payment that poses a critical market barrier because price resistance may limit market size.
Industry Perception of Science Base Readiness
There was a divergence of opinion among the pharmaceutical company interviewees about the readiness of the science base for cocaine pharmacotherapies. Representatives of two companies expressed skepticism about the readiness of the science base. One representative indicated that current limitations stem from a lack of understanding regarding the biological and genetic basis of addiction. A representative of a different company indicated that the current science base for achieving long-term efficacy for cocaine abuse and addiction is very weak. Furthermore, scientists from one company judged that the probability of a scientific breakthrough in the area of cocaine abuse and addiction in the near future is very low. In contrast, representatives of another pharmaceutical company indicated strongly that the science base is ready, and consequently that it is no longer a market barrier to development of cocaine pharmacotherapies. This company also reported that it had successfully identified several drug candidates that exhibited cocaine blocking activities in both in vivo and in vitro models. Scientific executives from one company who questioned the science base did suggest that there are opportunities for existing and potential products to be used as effective adjuncts to cocaine abuse therapy. For example, existing drugs for anxiety could help manage symptoms associated with withdrawal. The extent of company interviews was limited by the scope of this project.
Overcoming Critical Market Barriers
Any public policies intended to improve opportunities for developing pharmacotherapies for cocaine addiction must address the three critical barriers described here. It is not within the scope of this study to identify or analyze specific public policies to promote development or marketing of pharmacotherapies for substance abuse. Nevertheless, during the course of this study, certain types of strategies or initiatives emerged that would serve to lower these barriers and make the development of new pharmacotherapies for cocaine abuse more attractive to the pharmaceutical industry, as follows:
- Government funding of a considerable portion of new drug development
- Expansion and enhancement of the substance abuse treatment system
- Guaranteed market (e.g., purchase orders for minimum volumes of a medication)
- Extended market exclusivity (e.g., orphan drug or similar status)
The pertinence of such actions is supported by lessons from the case studies, suggestions raised by interviewees, and results of modeling diverse scenarios of new pharmacotherapy development described in this report. These strategies are consistent with certain of the strategies recommended elsewhere, e.g., certain ones raised by the IOM (1995), and merit further attention.
Government Funding of New Drug Development
In general, the investment to produce a new drug for a small market is no different than producing a new drug for a large market, but a company is less likely to recoup its investment in a small market. Government funding of new drug development can raise the science base and move any promising drug closer to launch. Improving the science base can increase opportunities to create new drugs, and increase the likelihood of producing drugs that will be more effective and acceptable to a wider market. Any drug that has progressed toward launch poses less risk of failure, and shortens the time to revenues, increasing the present value of the drug. From the standpoint of industry, this can shift the balance of risk and reward by effectively decreasing the front-end investment required for entering this risky market. Government funding may take the form of extramural and intramural research, cooperative research agreements with industry, or otherwise owning or acquiring the rights to promising compounds and then offering these to pharmaceutical companies willing to complete the development cycle, as in the "guaranteed handoff" scenario described in this report. Thus, government funding of new drug development could counteract the barrier of the small and uncertain market.
Expansion and Enhancement of Substance Abuse Treatment System
Improving the substance abuse treatment system can address a critical barrier to market access. Greater funding of treatment centers could increase the number of patients treated. It could also increase available spending per patient, enabling greater market penetration and more substantial prices for effective drugs. Requiring all substance abuse block grant recipients to offer approved pharmacotherapies would increase the scope of the market, particularly to the extent that this could overcome bias against pharmacological treatment of substance abuse. Assuring coverage and sufficient levels of reimbursement for appropriate use of pharmacotherapies could increase market size and ensure sufficient prices. State-of-the-art clinical practice guidelines rendered by expert panels and sponsored by authoritative organizations could become the standard of care. Clinical and payment policies could change accordingly, expanding the market for designated treatments. Thus, expansion and enhancement of the treatment system could help lower all three critical barriers.
Guaranteed Market
Through purchase orders or other means, government and/or other major payers could guarantee a substantial minimum market for a cocaine addiction pharmacotherapy. Guaranteeing a market in terms of number of patients and/or a number of drug units, a given price, and a specified period of time, would address directly the barrier of limited and uncertain payment for pharmacotherapy. Further, it could decrease uncertainty and improve the size of the market, as well as effectively expand the treatment system.
Extended Market Exclusivity
The prospect of splitting a small market with competitors may render a project financially infeasible. Provisions for market exclusivity via the existing Orphan Drug Act and the 1984 Drug Price and Competition and Patent Term Restoration Act, and/or similar means are necessary but not sufficient for lowering the barrier of a small and uncertain market. These provisions alone cannot expand the absolute size of the small market, but they can protect against competition in it and reduce uncertainty about market penetration. LAAM and naltrexone (as Trexan) have been granted orphan drug status for specified opiate indications. Broadly interpreting the Orphan Drug Act for substance abuse medications (e.g., the standard for designating orphan status to drugs intended to treat a condition affecting fewer than 200,000 people in the U.S.) or other policies giving similar market protections to drugs for this market could encourage companies to enter this market.
References
21 CFR 291 Drugs Used for Treatment of Narcotic Addicts.
Abt Associates. Rhodes W, Scheiman P, Pittayathikhum T, Collins L, and Tsarfaty V. Synthetic estimation applied to the prevalence of drug use. The Journal of Drug Issues. 1993;23(2): 297 - 321.
Agency for Health Care Policy and Research. Smoking cessation guideline technical report, number 18. National Technical Information Service, 1996. (NTIS No. PB97 - 104699).
Anderman B and Griffith RW. Clozapine-induced agranulocytosis: A situation report up to August 1976. Europ. J. Clin. Pharmacol. 1977;11: 199 - 201.
Behavioral Health Treatment. Naltrexone splits addiction community: awaiting more data, clinicians wonder it it's right to prescribe pills for alcoholism. Behavioral Health Treatment. 1996;1(4): 1-2.
Boodman SG. New hope for schizophrenia? The Washington Post. 16 February 1993; A10.
Bradford, A. President. Generico, Inc. Telephone interview. June 11, 1997.
Bradford, A. Telephone interview. June 17, 1997.
Brandeis University. Drug Services Research Survey: Phase I. Report to the National Institute on Drug Abuse. Waltham, Massachusetts, 1993a.
Brandeis University. Drug Services Research Survey: Final Report, Phase II. Report to the National Institute on Drug Abuse. Waltham, Massachusetts, 1993b.
CDC. Center for Disease Control's Tobacco Information and Prevention Source page. Statistics from the CDC's home page, http:/www.cdc.gov/nccdphp/osh.
Center for Substance Abuse Treatment. (1994) Approval and monitoring of narcotic treatment programs: a guide on the roles of federal and state agencies. Treatment Assistance Publication (TAP) Series #12.
Center for Substance Abuse Treatment. LAAM in the treatment of opiate addiction". Treatment Improvement Protocol (TIP) Series #22, 1995.
Crabtree BL. Review of naltrexone, a long-acting opiate antagonist. Drug Review. 1984; 3: 273 - 280.
Cruzan S. Clozapine approved for schizophrenia. Food and Drug Administration. 1989, Oct. 3. Press Release.
Cummings L. Personal Communications Lee Cummings, J.D., National Institute on Drug Abuse, August 8, 1997.
Demetrios J. NIDA's Naltrexone Research Program. NIDA Research Monograph. 1976;9: 5-11.
Denmead G., Fountain D., Harwood H, and Zhang D. Substance Abuse Treatment Services: Treatment Facilities and Funded Capacity. Report to the Office of Applied Studies, SAMHSA. Lewin-VHI, Inc., Fairfax, VA September, 1995.
Eissenberg, T. et al. Dose-related efficacy of levomethadyl acetate for treatment of opioid dependence: a randomized clinical trial. Journal of American Medical Association. 1997;277(24): 1945 - 1951.
Ereshefsky L, Wantanabe MD, Tran-Johnson TK. Drug review: clozapine: an atypical antipsychotic agent. Clinical Pharmacy. 1989;8: 691 - 709.
Everingham SC, and Rydell CP. (1994). Modeling the Demand for Cocaine. Drug Policy Research Center. RAND Corporation.
Feldman R., Fountain D, and Carney B. Demand for and Barriers to Use of LAAM: A Case Study of the Implementation of New Pharmacotherapies in the Treatment of Opiate Addiction. Report prepared for the Office of Science Policy, Legislation, and Education at NIDA, 1994.
Garrett HM. Ed. Red Book. Montvale, N.J., Medical Economics Data Production, 1994.
Gauen SE and Lee NL. Pharmacists' role in a smoking-cessation program at a managed health care organization. American Journal of Health Systems Pharmacology. 1995;52 (3): 294-6.
Geifler, R. President. Orpharm, Inc. Telephone interview. June 17, 1997.
Gerstein D. et al. Evaluating Recovery Services: The California Drug and Alcohol Treatment Assessment (CALDATA). Report to the State of California, Department of Alcohol and Drug Programs, Sacramento, CA, 1994.
Gerstein, D., and H. Harwood. eds. Treating Drug Problems. Volume 1: A Study of the Evolution, Effectiveness, and Financing of Public and Private drug treatment Systems, National Academy Press, 1990.
Hamill D. and Cooley P. National Estimates of Heroin Prevalence 1980-1987: Results from Analysis of DAWN Emergency Room Data. RTI Technical Report. Triangle Park, N.J.: Research Triangle Institute, 1990.
Harwood H, Thomson M, and Nesmith T. Healthcare Reform and Substance Abuse Treatment: The Cost of Financing Under Alternative Approaches. Fairfax, VA: Lewin-VHI, 1994.
Harwood, H., et al. How Many People are in Need of Treatment. Submitted to CSR, Inc. and the Office of National Drug Control Policy by Lewin-VHI, Inc. Fairfax, VA: Lewin-VHI, 1993.
Harwood H., Thomson M., Nesmith T. Healthcare Reform And Substance Abuse Treatment: The Cost of Financing Under Alternative Approaches. A Report to the Legal Action Center. Fairfax, VA: Lewin-VHI, Inc., February 1984.
Harwood HJ., et al. Economic Costs to Society of Alcohol and Drug Abuse and Mental Illness: 1980, submitted to the Alcohol, Drug Abuse, and Mental Health Administration, June 1984.
Haxby DG. Treatment of nicotine dependence. American Journal of Health Systems Pharmacology. 1995;52(3): 265-81.
Hebert JR, Kristeller J, Ockene JK, Landon J, Luippold R, Goldberg RJ, Kalan K. Patient characteristics and the effect of three physician-delivered smoking interventions. Preventive Medicine. 1992;21(5): 557-73.
Henry JA, Rivas CA. Constraints on antidepressant prescribing and principles of cost-effective antidepressant use. Pharmacoeconomics. 1997;11(5): 419-443.
Hirschfeld RMA, Keller MB, Panico S, Arons BS, Barlow D, Davidoff F, et. al. The National Depressive and Manic-Depressive Association consensus statement on the undertreatment of depression. JAMA; 1997;277(4): 333-340.
Homer JB.. A system dynamics model for cocaine prevalence estimation and trend projection. The Journal of Drug Issues. 1992;23(2): 251-279.
Hospital and Community Psychiatry. Sandoz guidelines for clozapine monitoring called too restrictive. Hospital and Community Psychiatry. 1991;42(5): 545.
IMS, Antidepressant Market Dollars by Channel, 1996.
Institute of Medicine. Fulco CE, Liverman CT, and Early LE. Development of medications for the treatment of opiate and cocaine addictions: issues for the government and the private sector. Committee to Study Medication Development and Research at the National Institute on Drug Abuse. Washington, D.C.: National Academy Press, 1995.
Institute of Medicine. Treating Drug Problems. Vol. I. Gerstein DR and Harwood HJ (Eds.). Committee for the Substance Abuse Coverage Study, Division of Health Care Services. Washington, D.C.: National Academy Press, 1990a.
Irvin SM. Treatment of depression with outpatient electroconvulsive therapy. AORN Journal. 1997; 65(3): 573-582.
Jaffe JH, Schuster CR, Smith BB, and Blachly PH. Comparison of acetylmethadol and methadone in the treatment of long-term heroin users: A pilot study. Journal of the American Medical Association. 1970;211: 1834-1836.
Johnson R, Gerstein D, Ghadialy R, and Gfroer J. Trends in the Incidence of Drug Use in the United States, 1919-1992. DHHS Publication No. (SMA) 96-3076, SAMHSA, Rockville, MD, 1996.
Johnston L. et al. National Survey Results on Drug Use from the Monitoring the Future Study, 1975 - 1993. National Institute on Drug Abuse (NIDA), Volume II, 1994.
Julius D. NIDA's Naltrexone Research Program. Nida Research Monograph. 1976;9: 5 - 11.
Kaim SC. Evolution of the National Academy of Sciences Study of Naltrexone. NIDA Research Monograph. 1976;9: 37-44.
Kunze RJ. Nothing Ventured: The Perils and Payoffs of the Great American Venture Capital Game. New York, NY: HarperBusiness: 1993.
Law M and Tang JL. An analysis of the effectiveness of interventions intended to help people stop smoking. Archives of Internal Medicine. 1995; 155 (18): 1933-41.
Lazowick AL. Managing patients with depression. Drug Topics Supplement. 1997; 40s-43s.
Martin WR, Jasinski DR, Mansky PA. Naltrexone, an antagonist for the treatment of heroin dependence. Effects in man. Arch Gen Psychiatry. 1977; 28: 784 - 91.
Martin WR and Jasinski DR. Characterization of EN-1639A. Clinical Pharmacol Ther. 1973;14: 142.
Medications Development Division. "Information on LAAM." Report prepared by the National Institute on Drug Abuse on the history of LAAM, 1994.
National Association of State Alcohol and Drug Abuse Directors (NASADAD) (Annual). State Resources and Services Related to Alcohol and Other Drug Problems. A Report to the Office of Applied Studies, Substance Abuse and Mental Health Services Administration, Rockville, MD.
Niaura R; Goldstein MG; Abrams DB. Matching high- and low-dependence smokers to self-help treatment with or without nicotine replacement. Preventive Medicine. 1994; 23 (1): 70-7.
Nunn-Thompson CL and Simon PA. Pharmacotherapy for smoking cessation. Clinical Pharmacology. 1989;8 (10): 710-20.
O'Brien CP, Greenstein R, Ternes J, Woody GE. Clinical pharmacology of narcotic antagonists. Annals of the NY Acad of Sciences. 1978;311: 232 - 240.
O'Brien CP, Volpicelli LA, Vopicelli JR. Naltrexone in the treatment of alcoholism: a clinical review. Alcohol. 1996;13 (1): 35 - 39.
O'Malley SS. Current strategies for the treatment of alcohol dependence in the United States. Drug and Alcohol Dependence. 1995;39 (Suppl. 1): S3 - S7.
Office of Applied Studies. Overview of the FY94 National Drug and Alcoholism Treatment Unit Survey (NDATUS) Data from 1993 and 1980-1993. Advance Report Number 9A. SAMHSA, Rockville, MD August, 1995.
Office of Applied Studies. National Drug and Alcoholism Treatment Unit Survey (NDATUS) Data for 1994 and 1980-1994. Advance Report Number 13. SAMHSA, Rockville, MD June, 1996.
Office of Applied Studies. National Household Survey on Drug Abuse: Population Estimates 1995. Substance Abuse and Mental Health Services Administration. US Department of Health and Human Services. Public Health Service, 1996.
Office of Applied Studies. Correspondence from Joan Epstein. Substance Abuse and Mental Health Services Administration. US Department of Health and Human Services. Public Health Service, 1997.
Office of Applied Studies. National Drug and Alcoholism Treatment Unit Survey (NDATUS) 191 Main Findings Report. DHHS Publication No. (SMA) 93-2007 SAMHSA, Rockville, MD, 1997.
Office of Applied Studies. National Admissions to Substance Abuse Treatment Services: The Treatment Episode Data Set (TEDS) 1992-1995. Advance Report Number 12, Treatment Episode Data Set. SAMHSA, Rockville, MD, February, 1997.
O'Malley SS. Strategies to maximize the efficacy of Naltrexone for alcohol dependence. NIDA Res Monogr. 1995;150: 53-64.
Oncology. Cancer pain remedy wins orphan drug status. Oncology. 1996;10(12): 1880.
Peck CC. FDA's position on the clozaril patient management system. Hospital and Community Psychiatry. 1990;41 (8): 876 - 877.
Pharmaceutical Approvals Monthly. New Drug Reviews: Abbott Serlect Schizophrenia Seven-Arm Study Could Meet FDA Standards for Comparative Labeling; Serlect will Compete Against Lilly's Zyprexa. Pharmaceutical Approvals Monthly. 1996;1(11).
Pink Sheet. 1994, September 5: 56 (36).
Pink Sheet. 1995, January 9: 57 (2).
Pink Sheet. 1996, March 18: 58 (12).
Pink Sheet. Clozaril active patient population has increased to 15,000, doubling since end of 1990. The Pink Sheet. 1991, Oct. 28: 53(43).
Pink Sheet. Marion Merrell Dow Cardizem line sales up 15% to $1.1. billion in 1992; Nicoderm contribute $27 million. The Pink Sheet. April 22, 1996, 4 (17).
Pink Sheet. Nicotine gum over-the-counter: only 12.4% of managed care plans would cover the product according to CHPG study. The Pink Sheet, October 9, 1995, 57(41).
Pink Sheet. SmithKline Beecham, Marion Merrell Dow combined OTC business has baseline sales of $660 million; venture provides critical mass, launching pad for switches. The Pink Sheet. August 24, 1992, 54 (34).
Plank SJ. Worksite smoking cessation using nicotine resin gum (Nicorette). American Journal of Public Health. 1987;77 (8): 1013-4.
Rawson, R. et al. (1996) A Three Year Progress Report on the Implementation of LAAM. Report written by researchers from the Friends Medical Science Research Center, Matrix Institute on Addictions, and the West Los Angeles VA Medications Development Research Unit.
Rawson, R. Friends Medical Science Research Center. Telephone interview. May 20, 1997.
Regier DA, Narrow WE, Rae DS, Manderscheid RW, Locke BZ and Goodwin FK. The de facto US mental and addictive disorder service system. Archives of General Psychiatry 1993;50.
Report of the National Research Council Committee on Clinical Evaluation of Narcotic Antagonists. Clinical evaluation of naltrexone treatment of opiate-dependent individuals. Arch Gen Psychiatry. 1978; 35: 335 - 40.
Resnick RB, Volavka J, Freeman AM et al. Studies of EN-1639A (naltrexone): a new narcotic antagonist. Am J Psychiatry. 1974; 131: 646 - 50.
Rhodes, W. Synthetic estimation applied to the prevalence of drug use. The Journal of Drug Issues. 1993;23(2), 297-321.
Rhodes W, Scheiman P, Pittayathikhum T, Collins L, and Tsarfaty V. What America's Users Spend on Illegal Drugs, 1988-1993. Executive Office of the President. Office of National Drug Control Policy, 1995.
Rice DP et al. The Economic Costs of Alcohol and Drug Abuse and Mental Illness: 1985, Report submitted to the Office of Financing and Coverage Policy of the Alcohol, Drug Abuse and Mental Health Administration, US Department of Health and Human Services, San Francisco, CA: Institute for Health and Aging, University of California, 1990.
Richards JW Jr . Cigarette smoking and Nicorette gum". Annals of Internal Medicine. 1987;106 (3): 482-3.
Sachs DP and Leischow SJ. Pharmacologic approaches to smoking cessation. Clinical Chest Medicine. 1991;12 (4): 769-91.
Schecter AJ, Friedman JG, Grossman DJ. Clinical use of naltrexone (EN-1639A): Part I: safety and efficacy in pilot studies. American Journal of Drug and Alcohol Abuse. 1974;1(2): 253 - 269.
Schecter AJ. The role of narcotic antagonists in the rehabilitation of opiate addicts: a review of naltrexone. American Journal of Drug and Alcohol Abuse. 1980;7(1): 1-18.
Scott-Levin, Source Prescription Audit 1996.
Scrip. Clozaril reaching 11,000 US patients and 48 states. Scrip. 1991, June 5: 1622: 14.
Scrip. Naltrexone approved for alcoholism. 1995, January 24: 23.
Scrip. Sandoz Pharma Sandimmun sales in 1993. Scrip. 1994, May 17: 1923: 17.
Sincich J. Director of Marketing. Roxane Laboratories, Inc. Telephone interview. June 27, 1997.
SmithKline Beecham. Nicorette Nicotine Gum to Quit Smoking, now Over the Counter. Information obtained from the Nicorette webpage created in 1997, http://www.nicorette.com.
Spangler JG; Salisbury PL. Smokeless tobacco: epidemiology, health effects and cessation strategies." American Family Physician. 1995;52 (5): 1421-30, 1433-4.
Suchinsky R. Associate Chief of Addicted Disorders, Department of Veteran's Affairs. Telephone interview. July 8, 1997.
Tan Sheet. In brief: Smoking cessation product sales. The Tan Sheet. November 18, 1996, 4 (47).
Tan Sheet. Nicorette 2 mg and 4 mg cleared for OTC use by FDA advisory committee. The Tan Sheet. October 2, 1995, 3 (40).
Tan Sheet. Nicorette actual-use studies support Waxman/Hatch exclusivity, SmithKline counsel argues. The Tan Sheet. April 15, 1996, 4 (16).
Tan Sheet. Nicorette labeling contraindicates use of nicotine patches. The Tan Sheet. February 19, 1996, 4(8).
Tan Sheet. Nicorette OTC use studies meet Waxman/Hatch exclusivity requirements, FDA says. The Tan Sheet. November 11, 1996, 4 (46).
Tan Sheet. Nicorette TV ads begin April 19, featuring "portraits" of quitter." The Tan Sheet. April 22, 1996, 4 (17).
Tan Sheet. OTC Nicorette will be available in retail stores by late spring following February 9 approval. The Tan Sheet. February 12, 1996, 4 (7).
Tan Sheet. OTC switch marketing exclusivity may be facilitated by 'paper trail'. The Tan Sheet. May 20, 1996, 4 (21).
Tan Sheet. Pharmacia & Upjohn Consumer Health sales down 17.8% to $153.2 million in first quarter. The Tan Sheet. May 5, 1997, 5 (18).
Tan Sheet. Pharmacia & Upjohn second quarter consumer health sales up 77% due to Nicorette and Rogaine. The Tan Sheet. August 12, 1996, 4 (33).
Tan Sheet. SmithKline Beecham Consumer takes over Nicorette marketing effective January 1. The Tan Sheet. August 23, 1993, 1 (26).
Tan Sheet. SmithKline Beecham worldwide OTC sales grow 11% to $1.03 billion in 1994, led by Tums. The Tan Sheet. February 27, 1995, 3 (9).
Tan Sheet. SmithKline Beecham's Nicorette/Nicoderm CQ generate $345 million in sales in 1996. The Tan Sheet. February 24, 1997, 5(8).
Tan Sheet. SmithKline forms partnership with American Lung Association to support Nicorette launch. The Tan Sheet. April 22, 1996, 4 (17).
Tan Sheet. SmithKline Nicorette education efforts include web site, Committed Quitters program. The Tan Sheet. April 22, 1996, 4 (17).
Tan Sheet. SmithKline Phase IV commitments include Nicorette study on use by adolescents. The Tan Sheet. February 19, 1996, 4 (8).
Tan Sheet. SmithKline worldwide OTC sales up 18% to $536 million in first quarter. The Tan Sheet. April 28, 1997, 5 (17).
Tan Sheet. Smoking cessation products' broader efficacy claims to be discussed by FDA advisory committee. The Tan Sheet. April 21, 1997, 5 (16).
Tokarski C. VA drops contract with drug maker. Modern Healthcare. 1990;July 30: 26.
United States Department of Health and Human Services. Smoking Deterrent Drug Products for Over-the-Counter Human Use; Establishment of a Monograph. Federal Register. 1982;47(2): 490-500.
US Congress. Senate. Committee on the Judiciary. Hard-Core Cocaine Addicts: Measuring-and Fighting-The Epidemic. A Staff Report Prepared for the Use of the Committee on the Judiciary, 1990.
Vocci F, Cummings L, and Montgomery A. Medications development division, National Institute of Drug Abuse. Interview. May 20, 1997.
Volpicelli JR, Volpicelli LA, O'Brien CP. Medical management of alcohol dependence: clinical use and limitations of naltrexone treatment. Alcohol & Alcoholism. 1996;30(6): 789 - 798.
Warner LA, Kessler RC, Hughes M, Anthony JC, and Nelson CB. Prevalence and correlates of drug use and dependence in the United States: results from the national comorbidity survey. Archives of General Psychiatry, 1995;52: 219-229.
Watson SJ, Akil H, Berger PA, Barchas JD. Some observations on the opiate peptides and schizophrenia. Arch Gen Psychiatry. 1979;36 (1).
Woodward A, Epstein J, Gfroerer J, Melnick D, Thoreson R, and Wilson D. (in Press). The Drug Abuse Treatment Gap: Recent Estimates. Office of Applied Studies, Substance Abuse and Mental Health Services Administration. U.S. Department of Health and Human Services.
Wright C. Personal Communications, Wright C, M.D., Food and Drug Administration, July 16, July 18, 1997.
Wright D, Gfroerer J, and Epstein J. The Use of External Data Sources and Ratio Estimation to Improve Estimates of Hardcore Drug Use from the NHSDA. Presentation at the Annual Meeting of the American Statistical Association, 1994.
Appendix A: Market Analysis Model
Drug development decisions on the part of pharmaceutical companies are based largely on determinations of risk-reward tradeoffs inherent in scientific and market factors. As described in this report, companies often have summary financial targets or hurdles that drive investment decisions. Two indicators of projected or actual financial performance that are often used in industry are net present value (NPV) and peak annual revenue (PAR). NPV is the difference between the present value of all cash inflows from a project and the present value of all cash outflows required for the investment, using an appropriate discount rate or required rate of return to calculate present values. PAR is the highest annual revenue achieved by a product during its market life.
Some of the main financial inputs to drug development decisions are: R&D costs; manufacturing, distribution, and marketing costs; opportunity cost of capital (or related parameters such as discount rate, interest rate, or required rate of return); sales revenues; and duration of the development and product/sales life. Factors that may not appear to have direct financial import, such as social stigma associated with a product, inclination of caregivers to consider pharmacological as opposed to behavioral interventions, or corporate commitment to further the greater societal good, do have financial implications that are considered by companies. The financial impacts of such factors can be estimated, if only at a rough level of approximation, and incorporated as such into decisions about pursuing projects.
This report uses a financial drug development model developed by The Lewin Group to quantify and portray basic relationships among such factors as market size, price, and revenue. The model is also used to simulate multiple, diverse scenarios of market conditions that could be faced by decision makers regarding development of new medications for treating cocaine abuse. Clearly, such decisions can be quite complex, and this model offers only a simplified quantitative tool for approximating relevant market conditions and outputs. Exhibit A-1 shows the input variables in the model.
| Exhibit A-1. Input Variables in the Market Model | |
|---|---|
|
Uncapitalized R&D costs |
Orphan drug status |
|
Stage of entry |
Years of orphan drug extension |
|
Discount rate |
Orphan drug tax advantage |
|
Wholesale price |
Years post-launch to competing drug |
|
Peak market size |
Years to replacement by competing drug |
|
Weeks of prescription |
First year MMDA* costs |
|
Expected peak prescriptions |
Duration of marketing campaign |
|
Years post-launch to peak prescriptions |
MMDA costs during marketing campaign |
|
Years to patent expiration |
MMDA costs after marketing campaign |
| *MMDA: manufacturing, marketing, distribution, administration | |
Each of these parameters can have a material impact on NPV and/or PAR.
Definitions of the model input variables are offered below. These include indications of the likely range of each variable, although the model allows entry of values outside of these ranges for most of these variables.
Uncapitalized R&D costs: Uncapitalized expenditures for R&D prior to marketing (range $50-250 million).
Estimates of the R&D expenditures required to bring a new drug from concept to market may vary widely, and are subject to multiple, complex, and often idiosyncratic conditions pertaining to a given product as well as various economic analyses and interpretations that are beyond the scope of this project. In its 1993 report, OTA estimated that the fully capitalized development cost of a medication approved by FDA in 1990 was as much as $359 million. Uncapitalized costs were about $135 million, spread over a 13-year development period. OTA also developed a hypothetical schedule of development costs over the development period, incorporating the preclinical and the clinical phases as well as the period to submit an NDA and gain FDA approval, as shown in Exhibit A-2. For any estimated level of uncapitalized R&D costs over a full development cycle, the model derives a distribution of costs over the development period based on the distribution of R&D costs from the OTA report. For any particular stage of entry (see below), the distribution of R&D costs is truncated up to that point.
| Exhibit A-2. Average Annual Cash Outlays for Development of a Medication, 1990 Dollars | |
|---|---|
|
Yr. 1: $5 million |
Yr. 8: $10 million |
|
Yr. 2: $10 million |
Yr. 9: $ 9 million |
|
Yr. 3: $15 million |
Yr. 10: $10 million |
|
Yr. 4: $17.5 million |
Yr. 11: $10 million |
|
Yr. 5: $17.5 million |
Yr. 12: $2.5 million |
|
Yr. 6: $15 million |
Yr. 13: $2.5 million |
|
Yr. 7: $11 million |
Total: $135 million |
| Source: Adapted from Office of Technology Assessment, 1993. | |
Stage of entry: Point in development cycle at which a company invests (range 0 to 13 years, or years corresponding to particular stages)
A company can choose to enter the product development cycle at various stages, including early research/preclinical, phase I trials, phase II, phase III, NDA submission, and at market approval. It is assumed that a typical product development cycle takes an average of 13 years. Each of the stages is associated with a particular average number of years to approval, i.e., 13 years at the beginning of the cycle, 9 years at the beginning of phase I trials, etc. The later a company enters the cycle (which may have progressed to that point with support of government or other research-based organizations or companies), the less is the assumed burden of development expenditures prior to any market approval.
Discount rate: The opportunity cost of capital applied to spending and revenues over the life of a product (range 10 to 20 percent annually).
The discount rate (or opportunity cost of capital or required rate of return) for the industry may vary from less than 10 percent to 20 percent or more depending upon financial conditions and other opportunities for investment relevant to companies. Riskier ventures tend to require higher rates than less risky ones. A typical rate used by large pharmaceutical companies is 12 percent.
Wholesale price per day: Average wholesale price per day of the medication (range $0.50 to $30.00).
Together with daily patients taking the medication this variable determines expected revenue to a pharmaceutical company. Pricing decisions by pharmaceutical companies are driven by a combination of factors including cost of development, cost of manufacturing, marketing and distribution, the existence of competing products or therapies, and perceived/actual price sensitivity of the market.
Peak market size: Expected peak daily number of patients taking the medication, i.e., patients enrolled in treatment and on a prescription on any given day (range 10,000 to 500,000).
This is a fundamental value driving this model. The alternative would be annual number of prescriptions written. When annual prescriptions written are combined with duration of the prescription (the next variable) it yields an estimate of daily patients taking the medication. Using daily patients as the parameter is preferred because there is better data about patients in treatment on a given day than there is on annual admissions to treatment (or the average duration of treatment, or the average duration of a course of medication). Generally, the more patients taking the medication, the higher will be revenue, given a stable price. It may take several years or more to achieve peak prescribing (another model parameter, below). Daily patients may be sensitive to price.
Weeks of prescription: Average time in weeks the patient will use/purchase the medication (range 4 to 52 weeks)
This factor is used with number of prescriptions made and the average dose to determine the total volume sold. For a medication for cocaine abuse, a likely prescription duration may be 3 months, which corresponds to the maximum duration of a course of naltrexone treatment for alcoholism. For opiate addiction, many patients are "maintained" indefinitely on methadone, and it is also possible to maintain patients on LAAM. A course of detoxification can be as brief as several days or last several months.
Years post-launch to peak prescriptions: This represents speed of product introduction and acceptance into the market (range 0 to 10 years).
The speed of product introduction can have an important impact on product profitability because of the limited patent life on medications, the threat of new medications entering the market, and the delay of achieving revenue from a product.
Expected peak prescriptions: Expected peak annual number of prescriptions written for the medication (derived from expected peak daily patients, and average weeks of taking prescription).
This number is determined by the number patients taking the medication on a given day, the duration of a course of medication, and the dose.
Years to patent expiration: Time in years from date of development decision until patent expires (range 0 years to 21 years).
Patent protection is critical to the profitability of a product, since generic companies tend to be very aggressive in pricing strategy. Patents are granted for nominally 17 years; companies may take a product into development for several years or more before applying for a patent, so that the time to patent expiration may exceed 21 years. Orphan drug status and certain forms of patent restoration may extend market exclusivity for a drug. Generic drug makers usually have far less need to recoup R&D investments, enabling major cost cutting relative to the price usually charged by the patent holder. Years of market exclusivity gained through orphan drug status run concurrently with any remaining years to patent expiration.
Orphan drug status: Whether or not a medication is expected to receive orphan drug status (1=yes, 0=no).
Orphan drug extension of market protection: Orphan drug status usually is granted effective at the time of FDA approval, and typically provides market exclusivity for 7 years.
Orphan drug tax advantage for development costs: Orphan drug status allows development costs to be expensed, effectively reducing development costs by 20 percent.
The Orphan Drug Act improves the economic incentives for development of medications to treat relatively rare disorders by first increasing the effective patent life for a drug granted orphan status, and second by allowing development costs to be expensed in the year incurred rather than requiring them to be capitalized and recouped against product revenues in the future. Years of market exclusivity gained through orphan drug status run concurrently with any remaining years to patent expiration. For a product whose patent expires, newly granted orphan drug status typically provides an additional 7 years of market exclusivity.
Years post-launch to introduction of competing drug: Expected years until introduction of competing product(s) (0 to 10 years).
Introduction of new products eliminates market exclusivity and reduces market penetration of the original product. Protection of market exclusivity (especially by patent or orphan drug status) delays the introduction of new products.
Years to replacement by competing drug: Expected market life of product after end of patent protection (0 to 10 years).
After market exclusivity expires, the market for a medication (i.e., for any given indication for that medication) typically erodes with the introduction of less-expensive generics and/or new competing therapies.
MMDA factors: Parameters pertaining to the duration and associated costs of manufacturing, marketing, distribution, and administration.
Companies make expenditures for manufacturing, marketing, product distribution, and administration over the life of a product. The model has parameters for first year MMDA costs as a ratio of first year revenue (range 1.0 to 2.0; typical value 1.2); duration of marketing campaign (typical value 5 years), MMDA costs during marketing campaign as a ratio of annual revenue (range 0.6 to 1.0; typical value 0.7), and MMDA costs after marketing campaign as ratio of annual revenue (range 0.2 to 1.0; typical value 0.5). For the purposes of simplifying the scenarios in this report, the "typical" MMDA factors shown here were held constant.
Appendix B: Summaries of Primary Studies on Need for Cocaine Treatment
ABT ASSOCIATES (Rhodes et al. 1993, 1995)
Rhodes, W. (1993). Synthetic Estimation Applied to the Prevalence of Drug Use. The Journal of Drug Issues 23(2), 297-321.
ESTIMATES
|
ABT ASSOCIATES (Rhodes et al., 1993) |
Estimate (in thousands) |
|---|---|
|
Cocaine |
|
|
Total(1990) |
2,036 |
|
At risk (not incarcerated)(1990) |
1,561 |
|
Heavy users in prison and jail(1990) |
214 |
|
Heavy users in high school(1990) |
24 |
|
High school age but incarcerated(1990) |
10 |
|
Heavy users who are high school dropouts(1990) |
7 |
|
Heavy users in college(1990) |
15 |
|
Additional heavy users in household(1990) |
205 |
|
ABT ASSOCIATES (Rhodes et al., 1993) |
Estimate (in thousands) |
|---|---|
|
Heroin |
|
|
Total(1990) |
665 |
|
At risk (not incarcerated) (1990) |
510 |
|
Heavy users in prison and jail(1990) |
142 |
|
Heavy users in high school(1990) |
6 |
|
High school age but incarcerated(1990) |
2 |
|
Heavy users who are high school dropouts(1990) |
2 |
|
Heavy users in college(1990) |
4 |
|
Additional heavy users in household(1990) |
0 |
DEFINITIONS
Rhodes estimates the number of weekly cocaine and heroin users. When necessary, the response more than 10 times per month was treated as weekly. The categories estimated are:
- At risk (not incarcerated)
- Heavy users in prison and jail
- Heavy users in high school
- High school age but incarcerated
- Heavy users who are high school dropouts
- Heavy users in college
- Additional heavy users in household
DATA SOURCES
Criminal justice involvement was estimated using arrests from the FBI's Uniform Crime Reports. Drug Use Forecasting (DUF) data were used to estimate the percentage of arrestees who would have tested positive for cocaine. Data from the Bureau of Justice Statistics (1990, 1991) were used to estimate the number of weekly cocaine users in prison. National Household Survey on Drug Abuse (NHSDA) data were supplemented with data from the High School Senior Survey to better estimate the number of high school students using cocaine. Also, data from the Bureau of Juvenile Statistics were used to complete the picture of high school-aged cocaine users. Data from the NHSDA prior to 1991 did not include college students who live in dormitories and fraternities. Consequently, they assumed that college students used drugs at the same frequency as high school seniors and made adjustments to the NHSDA data. They assumed that the homeless were largely included because of their involvement with the criminal justice system. Drug use in the military was assumed to be insignificant as was drug use in therapeutic communities. Their final estimates were deflated based on overlap across sources.
METHOD
Synthetic estimation is used to arrive at estimates of the numbers of weekly cocaine and heroin users for 1990 in various categories. Established relationships are used to infer drug use when direct measures are unavailable. Conceptually, cocaine and heroin users are comprised of intersecting groups of criminally involved persons, the homeless, high school students and drop-outs, college students, those in the military, those in residential treatment facilities, and those in households. The approach involves estimating the number of weekly users within each set, determining the overlap, summing across sets, and subtracting the overlap.
SOURCE
Rhodes, W., Scheiman, P., Pittayathikhum, T., Collins, L., and V. Tsarfaty. (1995). What America's Users Spend on Illegal Drugs, 1988-1993. Executive Office of the President. Office of National Drug Control Policy.
ESTIMATES
COMPOSITE ESTIMATES OF NHSDA AND DUF DATA
|
COCAINE |
1988* |
1989* |
1990* |
1991 |
1992 |
1993 |
|---|---|---|---|---|---|---|
|
Hard-core |
2540,525 |
2,624,312 |
2,468,509 |
2,218,700 |
2,339,381 |
2,127,166 |
|
Occasional |
7,347,000 |
6,465,843 |
5,584,686 |
5,440,415 |
4,330,521 |
4,054,117 |
|
HEROIN |
1988 |
1989 |
1990 |
1991 |
1992 |
1993 |
|
Hard-core |
591,990 |
607,046 |
533,630 |
465,305 |
444,372 |
496,309 |
|
Occasional |
539,000 |
504,446 |
469,891 |
368,102 |
289,557 |
229,251 |
| * The NHSDA estimates of cocaine users are adjusted for 1988 and 1990 to account for the survey's limited coverage during those years. | ||||||
DEFINITIONS
- Hard-core: defined in the NHSDA as one who uses cocaine at least 1 or 2 days a week every week for the year prior to the survey, or as one who used heroin on more than 10 days during the month prior to the survey.
- Hard-core: defined in the DUF as those who admitted using cocaine or heroin for more than 10 days during the month before being arrested.
- Occasional users: defined in NHSDA as those whose drug use was less than the criteria used for defining hard-core users.
- Occasional users: cannot be estimated from DUF.
DATA SOURCES
NHSDA supplemented with data from DUF and UCR.
METHOD
DUF data were converted to estimates of hard-core drug users throughout the criminal justice system. A weighting scheme was devised to estimate the number who would be expected to test positive in DUF sites. Since they over-represent large city lock-ups, a model was used to infer percentages who would have tested positive in non-DUF sites.
DATA ADJUSTMENTS
NHSDA data were adjusted for 1988 and 1990 to account for the survey's limited coverage during those years. The adjustment adds an estimate of hard-core drug users who live in college dormitories to the estimate of hard-core users derived from the NHSDA. Students living in college dormitories are represented in the 1991 and later NHSDA data. The NHSDA was not administered in 1989. Estimates for 1989 are the averages for 1988 and 1990.
Committee on Judiciary, U.S. Senate
SOURCE
U.S. Congress. Senate. Committee on the Judiciary. (1990). Hard-Core Cocaine Addicts: Measuring-and Fighting-The Epidemic. A Staff Report Prepared for the Use of the Committee on the Judiciary.
ESTIMATES
|
Estimate (in thousands) |
|
|---|---|
|
Committee on the Judiciary, U.S. Senate (1990) |
|
|
Total Unduplicated Hard-Core Cocaine Addicts |
2,200 |
|
Arrested (1988) |
1,530 |
|
Treated (1988) |
200 |
|
Homeless (1988) |
55 |
|
Household (1988) |
862 |
DEFINITIONS/CATEGORIES
Hard-Core Cocaine Addicts are defined as those who abuse cocaine at least once per week. This definition is intended to correspond to that of the NHDSA.
- Total Unduplicated Hard-Core Cocaine Addicts
- Arrested
- Treated
- Homeless
- Household
DATA SOURCES
Data on those receiving treatment were obtained by contacting the National Association of State alcohol and Drug Abuse Directors (NASADAD). The treatment data were supplemented with data from TOPS for 1979 to 1981. Criminal justice estimates were derived from DUF data. The NHSDA was the principal source for household data.
METHOD
Data from the individual states contained information on individuals receiving treatment for cocaine. From the TOPS data, it was estimated that 3 out of every 10 admissions were people who had been treated earlier. They therefore adjusted for multiple admissions. Furthermore, they assumed that 95 percent of admissions for cocaine were "hard-core" addicts, a conservative estimate according to the authors. They applied the overall estimate of hard-core addiction to a conservative estimate of the number of homeless to arrive at the number of homeless hard-core cocaine addicts. They used DUF data to estimate the drug use among arrestees. They applied the proportion testing positive for cocaine to the broader class of arrestees to arrive at the total number of cocaine users for the cities sampled. Then, they applied the average (47%) to cities not sampled. For small cities they assumed that 15% of arrestees would have tested positive for cocaine use. Finally, they adjusted based on overlap across data sources. They assumed that 40% of those arrested also sought treatment. They assumed that 10% of the homeless were not counted elsewhere. They left the number of arrestees intact because of adjustments from other sources. They concluded that 30% of arrestees were picked up by the household survey. Also, they assumed a 10% overlap between their treatment population and the NHSDA.
Homer (1993)
SOURCE
Homer, Jack B. (1993). A System Dynamics Model for Cocaine Prevalence Estimation and Trend Projection. In The Journal of Drug Issues 23(2), 251-279.
ESTIMATES
| ESTIMATES | |
|---|---|
|
Past Month Total |
3,600,000 |
|
Compulsive Prefer Crack(1990) |
1,296,000 |
|
Compulsive Prefer Powder(1990) |
576,000 |
|
Casual Prefer Crack(1990) |
1,008,000 |
|
Casual Prefer Powder(1990) |
720,000 |
DEFINITIONS
Broad Categories:
- used past month: used in NHSDA.
- used past year: used in NHSDA.
- used in lifetime: used in NHSDA.
Sub-categories within each of the Broad Categories:
- Compulsive, prefer crack.
- Compulsive, prefer powder.
- Casual, prefer crack.
- Casual, prefer powder.
- Compulsive (heavy) refers to users who have used every week for the past year (average of 8 grams per month). Corresponds to NHSDA definition of weekly use.
- Casual (light) users average one-half gram per month.
DATA SOURCES
NHSDA data for the years 1976, 1977, 1979, 1982, 1985, 1988, and 1990 were supplemented with data from the High School Senior Survey (HSSS) annual reports 1976-1990, Drug Abuse Warning Network (DAWN) 1976-1989, the Drug Use Forecasting (DUF) 1988-1989, Uniform Crime Reports (UCR) 1977-1989, Offender-Based Transaction Statistics (OBTS) 1983-1987, National Narcotics Intelligence Consumers Committee (NNICC) Reports 1977-1989, and the System to Retrieve Drug Evidence (STRIDE) 1977-1990.
METHOD
A system dynamics model was developed to generate estimates and projections on the national prevalence of cocaine use. Casual hypotheses/relationships were modeled or translated into equations and attempts to explain historical data resulting in the rejection of some models and refinement of others. Emphasis was placed on modeling endogenous (feedback) relationships and internal factors rather than exogenous (external) influences (model diagram is pictured on page 259 of article). Endogenous factors/variables included cocaine user population (dependent variable(s)), reported cocaine use prevalence, social exposure to cocaine, perceived health risks, morbidity and mortality, perceived legal risks, drug law incarceration and arrests, and several factors related to the cocaine market. Exogenous factors/variables included marijuana use prevalence, introduction of crack cocaine, seizure fraction, and arrest rate and incarceration fraction.
Institute of Medicine
SOURCE
Gerstein, D., and H. Harwood. eds., 1990. Treating Drug Problems. Volume 1: A Study of the Evolution, Effectiveness, and Financing of Public and Private Drug Treatment Systems, National Academy Press.
ESTIMATES
|
IOM METHODOLOGY (Gerstein and Harwood 1990) |
Numbers (in thousands) |
|---|---|
|
(Harwood et al., 1993) Total Estimate of Treatment Need(1991) |
4,887 |
|
Total Estimate of Treatment Need(1987-1988) |
5,455 |
|
Household Population: Clear need (1987-1988) |
1,500 |
|
Household Population: Probable need (1987-1988) |
3,100 |
|
Homeless(sheltered. street, and transient) |
170 |
|
Correctional Custody |
320 |
|
Probation and parole |
730 |
|
Pregnancies (live births) |
105 |
|
Less overlaps |
-470 |
Need for Treatment:
Estimates from the NHSDA based on a combination of:
- frequency and intensity of drug use;
- number of symptoms of dependence in past year reported in NHSDA; and
- number of problems from drug use in past year reported in NHSDA.
All those using their "primary" drug on 9 or more days per month were classified as in clear or probable need for treatment, based on whether they self-reported 3 or more, or fewer symptoms/problems, respectively. Use of a primary drug 2 to 8 days per month plus 3 or more symptoms/problems were equivalent to probable need for treatment.
Estimates for other populations and surveys based on measures of frequency/intensity of use and/or reported symptoms/problems associated with drug use that are indicative of a need for treatment. The various surveys use quite different definitions and items.
DEFINITIONS
- Clear need was defined in terms of exceeding thresholds on the following 3 criteria: illicit drug consumption at least three times weekly, at least one explicit symptom of dependence, and at least one other kind of functional problem attributed to drug use.
- Probable need was assigned if level of consumption, number of symptoms, or number of problems fell below one threshold value but exceed the threshold on others.
- Possible need was assigned if there was at least some monthly use and some indication of symptoms or problems.
- Unlikely need was assigned to all others.
DATA SOURCES AND METHOD
The principal source of data was the National Household Survey of Drug Abuse (NHSDA). DUF data were used to estimate the need for treatment among arrestees with extrapolations being used to extend the estimates to the national level. Estimates based on State prison surveys for 1986 revealed that 43 percent were in need to treatment according to the criteria for drug dependence. This percentage was applied to the 1987 state and federal prison census for 1987. The same percentage was applied to the parolees population. The homeless population estimates of prevalence rates ranged from 10 to 33.5 percent. The median value (20%) was applied to the estimate of the homeless population to derive the number in need to treatment. The 1988 NIDA survey provided an estimate of the number of women in high fertility age brackets who used illicit drugs. The overall birth rate for that age group was applied and an estimate of the number of fetal exposures was generated.
National Comorbidity Survey
SOURCE
Warner, L.A., Kessler, R.C., Hughes, M., Anthony, J.C., and C.B. Nelson (1995). Prevalence and Correlates of Drug Use and Dependence in the United States: Results from the National Comorbidity Survey. Archives of General Psychiatry, Vol. 52: 219-229.
National Comorbidity Survey
- Past 12 month drug use meeting the DSM-III-R criteria for drug dependence or abuse in a national probability sample of 10,000 individuals.
Office of Applied Studies/SAMHSA
SOURCE
Woodward, A., Epstein, J., Gfroerer, J., Melnick, D., Thoreson, R., and D. Wilson. (in press). The Drug Abuse Treatment Gap: Recent Estimates. Office of Applied Studies, Substance Abuse and Mental Health Services Administration. U.S. Department of Health and Human Services.
ESTIMATES
|
SAMHSA (Woodward et al., in press) |
Estimate (in thousands) |
|---|---|
|
Total |
7,100 |
|
Level 1 Need(1994) |
3,500 |
|
Level 2 Need(1994) |
3,600 |
DEFINITIONS
Total Treatment Need is defined as someone meeting at least one of the following four conditions:
1. Drug Dependence: A person is defined as dependent for a specific drug in 1995 if they indicate that they have used that drug in both the core and the dependence sections of the questionnaire and they meet 3 of the 6 DSM-IV criteria by the 1995 dependence questions.
2. Heavy Drug Use. Any of the following in the past year:
a. Used heroin at least once in the past year.
b. Used marijuana daily.
c. Frequent use (52 + days/weekly) of some other illicit drug.
3. IV Drug Use: Used heroin, cocaine or stimulants with a needle in the past year.
4. Treated for Drug Abuse: Received treatment for any illicit drug in the past year.
Level 2 Treatment Need is defined as a person with at least one of the following four conditions:
1. Drug Dependence: dependence on any illicit drug except marijuana in the past year.
2. Heavy Drug Use: Any of the following in the past year:
a. Used heroin at least once in the past year.
b. Used marijuana daily AND dependent on marijuana.
c. Frequent use (52 + days/weekly) of some other illicit drug
d. Daily use of any illicit drug except marijuana
3. IV Drug Use: Used heroin, cocaine or stimulants with a needle in the past year.
- Treated for Drug Abuse: Received treatment for any illicit drug at a specialty facility
in the past year.
Level 1 Treatment Need is defined as those meeting the conditions for total treatment but not meeting the criteria for Level 2 treatment need.
DATA SOURCES
The principal source of data is NHSDA data with ratio estimation to account for under-reporting and undercoverage. Supplemental data sources include National Drug and Alcohol Treatment Unit Survey (NDATUS) now called the Uniform Facility Data Set, the Drug Services Research Survey (DSRS), and the Uniform Crime Report (UCR).
METHOD
The National Household Survey of Drug Abuse covers non-institutionalized populations aged 12 and over. The sampling frame under-estimates drug abusers because it does not cover institutionalized populations, the homeless, and those in treatment. Also, adjustments for under-reporting are likely to be an issue. The authors use ratio estimation and data from supplemental sources to adjust the NHSDA estimates of substance abuse. Ratio estimation is built on the idea that better estimates are possible if there is a known population estimate of a related variable. The ratio estimation procedure uses both the in-treatment and arrest counts to obtain corrected national counts of 1) those arrested and treated, 2) treated but not arrested, 3) arrested but not treated, and 4) not arrested and not treated. The adjusted estimates of total treatment need, and Level I and Level II need are presented above.
Older OAS/SAMHSA definitions:
The DEP definition attempts to approximate the DSM-III-R definition.
The DSM-III-R defines a person as dependent if they meet 3 of 9 criteria for dependence.
The NHDSA survey contains questions that are combined to approximate 5 of the 9 DSM categories. They include: tolerance, withdrawal, inability to stop or control substance use, giving up or reducing social occupational or recreational activities, and continued substance use despite knowing consequences.
The DEP definition classifies someone as dependent if they respond positively to 2 of the 5 items.
RAND Corp.
SOURCE
Everingham, S.C., and C. P. Rydell 1994. Modeling the Demand for Cocaine. Drug Policy Research Center. RAND Corporation.
ESTIMATES
| ESTIMATES | |
|---|---|
|
Heavy Cocaine Users (1992) |
1.72 million |
|
Light Cocaine Users (1992) |
5.60 million |
|
US Cocaine Users Observed (1991) |
7.27 million |
|
US Cocaine Users Modeled (1991) |
8.02 million |
DEFINITIONS
- Heavy users: people who used cocaine weekly over the course of the last year (NHSDA).
- Light users: all others.
DATA SOURCES
NHSDA data were supplemented with data from the Housing and Urban Development (HUD), National Bureau of Economic Research, ICF, and 1986 Survey of Inmates of State Correctional Facilities.
METHOD
A Markovian two-state, four parameter model was used. The two states were heavy and light users. The four parameters were 1) light users to non-users, 2) light users to heavy users, 3) heavy users back to light users, and 4) heavy users that flow out of cocaine use. The numbers of light and heavy users were year-dependent.
DATA ADJUSTMENTS
Estimates were based on the NHSDA and supplemented with data to account for under-counting of the homeless and incarcerated. Therefore, while they adjust for incarcerated and homeless populations, they make no adjustment for under-reporting.
Appendix C: Need For Substance Abuse Treatment
Table: Need for Cocaine Treatment
| Source | Estimate |
|---|---|
| ABT ASSOCIATES (Rhodes et al., 1995) | |
| Hardcore (1993) | 2,127,000 |
| ABT ASSOCIATES (Rhodes et al., 1993) | |
| Total (1990) | 2,036,000 |
| At risk (not incarcerated) (1990) | 1,561,000 |
| Heavy users in prison and jail(1990) | 214,000 |
| Heavy users in high school (1990) | ALIGN="RIGHT"> 24,000 |
| High school age but incarcerated (1990) | 10,000 |
| Heavy users who are high school dropouts (1990) | 7,000 |
| Heavy users in college (1990) | 15,000 |
| Additional heavy users in household (1990) | 205,000 |
| RAND | |
| Heavy users (1992) | 1,720,000 |
| HOMER (1993) | |
| Past Month Total | 1,872,000 |
| Compulsive Prefer Crack (1990) | 1,296,000 |
| Compulsive Prefer Powder (1990) | 576,000 |
| SAMHSA (Office of Applied Studies, unpublished data, 1997) | |
| Past Year Cocaine Users in Need of Treatment (1995) | 2,703,000 |
| Level 1 (1995) | 567,000 |
| Level 2 (1995) | 2,136,000 |
| Committee on the Judiciary US Senate (1990) | |
| Total Unduplicated Hard-Core Cocaine Addicts | 2,200,000 |
| Arrested (1988) | 1,530,000 |
| Treated (1988) | 200,000 |
| Homeless (1988) | 55,000 |
| Household (1988) | 862,000 |
Table: Need for Heroin Treatment
| Source | Estimate |
|---|---|
| ABT ASSOCIATES (Rhodes et al., 1995) | |
| Hard core (1993) | 496,000 |
| ABT ASSOCIATES (Rhodes et al., 1993) | |
| Total(1990) | 665,000 |
| At risk (not incarcerated) (1990) | 510,000 |
| Heavy users in prison and jail(1990) | 142,000 |
| Heavy users in high school(1990) | 6,000 |
| High school age but incarcerated(1990) | 2,000 |
| Heavy users who are high school dropouts(1990) | 2,000 |
| Heavy users in college(1990) | 4,000 |
| Additional heavy users in household(1990) | 0 |
| SAMHSA (OAS, 1996) | |
| Used at least once in lifetime (NHSDA, 1995) | 2,400,000 |
| Used at least once in lifetime (NHSDA, 1995) with Adjustments for Under-reporting and Under-coverage | 2,900,000 |
| Used at least once in past year (NHSDA, 1995) | 428,000 |
| Used at least once in past year (NHSDA, 1995) with Adjustments for Under-reporting and Under-coverage | 541,000 |
| Hamill and Cooley (1990) | |
| Number of heroin addicts(1987) | 640,000 -1,065,000 |
Source: Providers, The National Specialty Substance Abuse Treatment System
Appendix D: Literature Search Strategies For Case Study Reports Of LAAM, Naltrexone, Clozapine, And Nicorette
Literature Search Methods for LAAM Case Study
| Database Type | Database Name | Years | MeSH |
|---|---|---|---|
| NLM | MEDLINE | 1966-Present | LAAM and EXP opiate addiction LAAM, Methadone, and clinical trial (pt) |
| HSTAR75 | 1975 - 1984 | LAAM | |
| HSTAR | 1984-Present | LAAM | |
| Dialog | FDA Pink Sheet | 1987 - Present | LAAM and opiate addiction |
| FDA Tan Sheet | 1990 - Present | LAAM and opiate addiction | |
| Health and Wellness Database | 1976-1997 | LAAM and opiate addiction |
Literature Search Methods for Naltrexone Case Study
| Database Type | Database Name | Years | MeSH |
|---|---|---|---|
| NLM | MEDLINE | 1980-Present | Naltrexone and EXP alcoholism ALIGN="JUSTIFY"> Naltrexone and EXP Drug Industry |
| 1966-1984 | trexan and review (pt) trexan and clinical trial (pt) | ||
| HSTAR75 | 1975 - 1984 | Naltrexone | |
| HSTAR | 1984-Present | Naltrexone | |
| HSRProj | ????-Present | Naltrexone (5) | |
| Dialog | FDA Pink Sheet | 1987 - Present | Naltrexone |
| Scrips | 1980 - Present | Naltrexone and Alcohol | |
| Health and Wellness Database | 1976-1997 | Naltrexone and Alcohol | |
| IAC Trade and Industry Database | 1976-1977 | Naltrexone and Alcohol | |
| ABI/INFORM ® | 1971-1997 | Naltrexone and Alcohol |
Literature Search Methods for Clozapine Case Study
| Database Type | Database Name | Years | MeSH |
|---|---|---|---|
| NLM | MEDLINE | 1980-Present | Clozapine and Drug Costs or Drug Industry Clozapine and Review (pt) Clozapine and Clinical Trial (pt) |
| 1966-1984 | Clozapine and Review (pt) Clozapine and Clinical Trial (pt) | ||
| HSTAR75 | 1975 - 1984 | Clozapine | |
| HSTAR | 1984-Present | Clozapine | |
| HSRProj | ????-Present | Clozapine | |
| Dialog | FDA Pink Sheet | 1987 - Present | Clozapine and Schizophrenia |
| Scrips | 1980 - Present | Clozapine and Schizophrenia | |
| Health and Wellness Database | 1976-1997 | Clozapine and Schizophrenia | |
| IAC Trade and Industry Database | 1976-1977 | Clozapine and Schizophrenia | |
| ABI/INFORM ® | 1971-1997 | Clozapine and Schizophrenia |
Literature Search Methods for Nicorette Case Study
| Database Type | Database Name | Years | MeSH |
|---|---|---|---|
| NLM | MEDLINE | 1966-Present | Nicorette and nicotine addiction Nicorette and smoking cessation therapy Nicotine polacrilex |
| HSTAR75 | 1975 - 1984 | Nicorette and nicotine addiction Nicorette and smoking cessation therapy | |
| HSTAR | 1984-Present | Nicorette and nicotine addiction Nicorette and smoking cessation therapy | |
| Dialog | FDA Pink Sheet | 1987 - Present | Nicorette and smoking cessation therapy |
| FDA Tan Sheet | 1990 - Present | Nicorette and smoking cessation therapy | |
| Health and Wellness Database | 1976-1997 | Nicorette and smoking cessation therapy |
Appendix E: Selection Criteria And Discussion Guide For Interviews With Private Firms
Interviews With Private Firms
Pursuant to Task 6.0 of our contract, the present document contains the three components of the interviews with private firms: a) selection criteria for firms to be interviewed, b) list of selected firms to be interviewed, and c) discussion guide for interviews. The purpose of meeting with private firms is to explore and characterize private industry's views on market barriers to the development of these pharmacotherapies and on the readiness of the science base for developing such medications. We will meet with leaders (e.g., senior executives, marketing and research directors) of pharmaceutical and venture capital firms (a total of five firms), one firm at a time.
We will seek industry views on market barriers with the objective of determining their relative impacts on private investment in these pharmacotherapies. In addition, we will determine the facts and reasoning underlying industry's judgment on these issues, and the extent to which additional information, such as that obtained in our market analysis, might alter industry's view (or the view of one or more firms) on a particular market barrier. Finally, we will determine the industry's perceptions about the readiness of the science base for the development of such pharmacotherapies.
Selection Criteria for Firms and Candidate List
As discussed with ASPE informally and during our meeting of May 1, we will select firms that are actively working, or appear to be fully capable of initiating efforts, in the field of substance abuse. We will select firms that may be involved in such efforts and, to the extent possible, choose firms that can provide a diverse set of experiences and perspectives on drug development. In addition, because of potential scheduling difficulties, we will select firms that are most likely to cooperate in the study. Below, we list the types of companies we propose to target.
- A large pharmaceutical company that is fully capable of developing or licensing a product in the treatment of addiction. We would seek a company that is likely to share insights into its strategies and plans.
- A mid-size to large pharmaceutical company with a strong CNS identity and/or mission.
- A small pharmaceutical company that is developing at least one product relevant to the present study.
- Two venture capital / investment banking firms with real or potential interest in drug development.
Of course, there is no assurance that a given company would be willing to participate in the study. In addition, scheduling may be difficult, given that our interviews must be with senior individuals. We would therefore propose to initiate calls to first-choice companies as soon as possible to discuss participation and to obtain commitments.
Discussion Guide
The discussion guide is designed to ensure that questions addressing private sector views on market barriers to the development of cocaine pharmacotherapies and the readiness of the science base in developing such medications are appropriately covered.
Our interviews will be designed to assess the primary concepts of concern to ASPE. For example, we will determine whether any stigma associated with management of drug addiction plays a role in the decision to develop a compound, as do more conventional factors such as market size and anticipated return on investment (ROI). We will also probe on the extent to which a pharmaceutical company's decision to proceed with a compound is based on potential market size in absolute terms, or on market size relative to other factors (e.g., potential for orphan drug status). We will also seek insights regarding interactions between research and marketing departments within a pharmaceutical company concerning assessment of a compound's market potential. Some of the questions posed to venture capital executives will differ from those posed to pharmaceutical company officials.
Wherever possible, we will ask interviewees to respond in the context of particular case examples, including products that are now or had been considered for development, products in R&D pipelines, and products on the market. We recognize that responses to various questions about factors influencing decisions to pursue pharmacotherapies may be product- and application-specific, i.e., with respect to readiness of the science base and various types of market barriers.
General Company Background (for Rx companies only)*
- What is the size of the company (e.g., employees, annual revenues)?
- How geographically diverse is your market?
- Please describe your company's portfolio in health care and, in particular, in the area of CNS-related products.
- Do you have any products, in the pipeline or on the market, in the area of substance abuse addiction, or do you have relationships with companies that do have such products?
*Some of this information may be available in company reports.
Approach to evaluation of new investments (for Rx companies only)
- What current priorities do you have in health care, and, in particular, in the area of CNS-related products?
- What criteria do you use to establish these priorities?
- Who is responsible for the evaluation of new investments in health care?
- What are the most important factors in deciding whether or not to pursue the development of a drug (e.g., ROI, focus in particular disease area, corporate mission, other competitive factors)?
- Are there particular financial thresholds that serve as criteria for pursuing a drug, e.g., a particular ROI or net present value (NPV)?
- Do you have internal staff capacity devoted to conducting market analyses? If so, how many FTE employees, and how is this organized?
- Do you have internal staff capacity for regulatory affairs?
- What are the relative roles (and authorities) of the research department and the marketing department in the decision to pursue the development of a drug?
- Have you conducted any market analyses in the area of cocaine abuse and addiction?
- Do you consider market analyses of other drugs (internal or external) when considering markets for treatments for cocaine abuse and addiction?
Approach to evaluation of new investments (for venture capital firms only)
- What current priorities do you have in health care, and, in particular, in the area of CNS-related products?
- What criteria do you use to establish these priorities?
- What are the most important factors in deciding whether to invest in a drug and, if so, when to invest (e.g., potential market size, market penetration, total capital required, break-even point, ROI or NPV threshold, regulatory status, product cycle)?
- What due diligence do you perform on drugs that you are evaluating for potential investment? Can you briefly describe this process?
- Have you invested in or evaluated investment opportunities in the area of pharmacotherapies for cocaine abuse and addiction? If yes, for which drug, and why?
Perceived Market Barriers (for both Rx and venture capital firms)
- What do you perceive to be the most important market barriers to developing pharmacotherapies for addiction?
- In addition to the ones you have cited, which of the following factors do you consider to be important market barriers? (This is a sample list meant to complement those barriers identified above and to help elicit further insights; it may not be necessary to review all of these with each interviewee.)
- size of the market
- cost of developing and marketing a drug
- pharmacological feasibility
- potential for clinical value
- public sector role in development and/or evaluation
- FDA requirements affecting research, development, and approval
- DEA requirements affecting research, development, and approval (esp. implications of treatments with addictive vs. non-addictive properties)
- other requirements for development and approval (e.g., state requirements)
- clinical trials management (i.e., enrollment and retention issues, clarity of definable clinical endpoints)
- time frame for development and approval
- patent life / protection
- orphan drug requirements
- price potential for the drug
- coverage / reimbursement requirements
- dispensing requirements (e.g., prescription restrictions, clinical settings)
- social attitudes about treatments for substance abuse (including, e.g., stigma that may prevent firm from exploring substance abuse indications for drugs marketed for other conditions)
- physician attitudes about treatments for substance abuse
- commercial / competitive value
- probability of achieving marketing success
- legal considerations
- Among these real or potential barriers, which are of the greatest and least consequence from industry's perspective?
- Would the lowering or elimination of one or more of these barriers significantly increase the magnitude of industry activity in pharmacological treatment for cocaine abuse and addiction?
- Does the venture capital world hold a different view?
Readiness of Science Base (for Rx companies only)
- Is the readiness of the science base currently a barrier to the development of cocaine pharmacotherapies? If so, why, and specifically in which areas (e.g., craving control, relapse prevention)? What is the basis for your assessment of the science base (e.g., internal strategic assessments)?
- Is the infrastructure (including in the public and private sectors) for clinical research adequate for strengthening the science base? What forms the basis for your answer?
- Would a seminal discovery be required to stimulate private sector interest? What forms the basis for your answer?
- How would you compare the state of the science base in this field to other fields of pharmaceutical R&D? What forms the basis for your answer?
- What is the maturity or readiness of the science base in this field compared to other fields, e.g., HIV/AIDS research in the late 1980s, Alzheimer's disease research today?
- Does the science base for anti-addiction drugs have applications for other products in other areas? What forms the basis for your answer?
- Does the current science base for CNS drugs offer potential for development of effective new cocaine pharmacotherapies? Can you give some examples?
- What drugs for treating cocaine abuse and addiction are in the pipeline now (to the extent this information can be made available)?